
A Multidisciplinary Approach to the Treatment of Anti-NMDA-Receptor Antibody Encephalitis: A Case and Review of the Literature
Abstract
Anti-NMDAR (N-methyl-d-aspartate receptor) encephalitis is a novel autoimmune and paraneoplastic disease often presenting as acute psychosis. Few studies exist in the psychiatric literature on neuroimmunity and behavioral management. This article reviews the epidemiology, diagnosis, pathophysiology, and management of this disease from a neuropsychiatric perspective. Patients have potential for near-complete recovery with early diagnosis and intervention. In addition to immune-suppression and tumor removal, electroconvulsive therapy may be an important tool in treatment of the underlying process in cases developing life-threatening catatonia. Psychiatrists should be familiar with treatment options, since they may be consulted within the context of a multispecialty team.
First described in 2007 by Dalmau et al., anti-NMDAR (N-methyl-d-aspartate receptor) encephalitis has been recently recognized as another cause of autoimmune and paraneoplastic limbic encephalitis (LE).1,2 Although primarily a paraneoplastic syndrome of young women with ovarian teratomas, an increasing number of cases have been reported in both men and women, ranging in age from 23 months to 76 years, with and without tumors.3,4 Cases have presented to neurologists, psychiatrists, and emergency physicians. They eventually call upon the expertise of a multidisciplinary team.
Although over 400 cases have been identified by Dalmau and his colleagues, there are few studies reported in the psychiatric literature on neuroimmune and behavioral management, especially in adolescents. In order to demonstrate the complexity of this disease and its clinical management, we have included the following description of an case in an adolescent.
Case Presentation
A previously-healthy 14-year-old African American girl reported a weeping blister below her nose, a sunburn-like facial rash, headache, and a temperature of 37.8°C (100°F). These findings quickly resolved without intervention. Two days later, she developed acute-onset auditory hallucinations and paranoia. She told her parents, “God said everything will be OK.” She was brought to the emergency department after becoming physically threatening. The patient’s past psychiatric and medical history were unremarkable.
After spending 2 days in an inpatient psychiatric unit, she was transferred to the pediatric intensive-care unit for further work-up. She demonstrated hyperactivity, irritability, increased speech, insomnia, and continued paranoia. Her CSF revealed a lymphocytic pleocytosis of 49 nucleated cells per mm3 with 98% lymphocytes and normal glucose and protein concentrations. The patient was started empirically on vancomycin, ceftriaxone, and acyclovir. The antibiotics were discontinued when her blood and CSF cultures returned negative. She was continued on acyclovir for 21 days to cover for presumed viral encephalitis of unknown etiology.
Her head CT, basic metabolic panel, and urine toxicology screen were unremarkable. She experienced one witnessed tonic–clonic seizure, after which she was placed on prophylactic levetiracetam. Her electroencephalogram (EEG) revealed frontal slowing, but no epileptiform activity.
A week later, the patient’s cognitive and neurological status deteriorated significantly. She became incontinent of bowel and bladder, and refused to take anything by mouth, necessitating nutritional support by nasogastric tube. Her mental status waxed and waned; at times she produced speech, with word-finding difficulty, and at others, she remained mute. For periodic agitation, lorazepam 1 mg iv prn provided some relief.
Given a broad differential, including endocrine, infectious, inflammatory, and autoimmune etiologies, an extensive medical work-up ensued, but was unrevealing. Her creatinine kinase was elevated, at 1,116 IU/L. Standard autoimmune antibodies were not detected in her CSF or serum. Her CSF was also negative for voltage-gated potassium-channel antibodies. A series of other tests for viral and parasitic etiologies were negative. She was started on a course of prednisolone 30 mg by nasogastric tube every 12 hours for presumed autoimmune encephalitis.
Six weeks after the initial onset of symptoms, her CSF was found to be positive for anti-NMDAR antibodies (Laboratory of Neuro-Oncology, University of Pennsylvania). Full-body MRI and PET scan were negative for malignancy. A 3-day course of intravenous immunoglobulin (IVIG) was administered, without significant improvement.
Two months after her initial symptoms, her CSF showed no WBCs, but remained positive for anti-NMDAR antibody. Serum ESR and CRP were found to be elevated, at 114 mm/sec and 39 mg/L respectively.
The patient’s mental status varied in terms of alertness. She demonstrated periods of dystonia, dyskinesia, spastic rigidity, cogwheel rigidity, bilateral upper-extremity hyperreflexia, as well as periods of decreased tone in all extremities. She made infrequent, unintelligable verbal communication with family members, but was able to recognize herself and her family in photographs. The patient remained incontinent of bowel and bladder function and required nutrition by nasogastric tube.
A psychiatric evaluation was obtained for recommendations on continued aggressive behavior and impulsivity. The patient showed limited improvement in aggression after a 1-week trial of oral risperidone 1 mg ng q8hr prn, and some improvement in agitation and insomnia with diphenhydramine 25 mg iv q6hr prn. Around this time, the neurology team recommended carbidopa-levodopa 10 mg/100 mg bid for muscle rigidity.
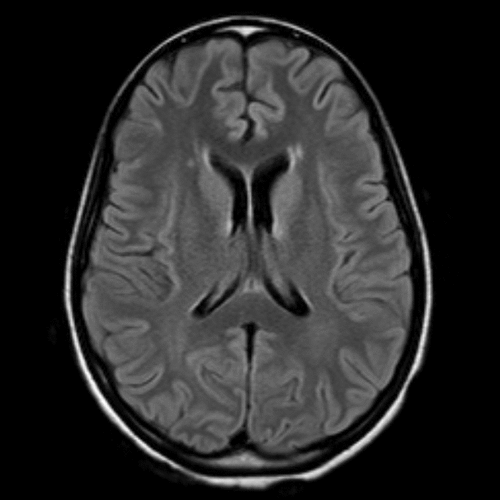
Seven weeks after symptom onset, a second brain MRI revealed minimal nonspecific periventricular white-matter changes (Figure 1). A repeat EEG showed a background with moderately organized waves of 10 per second. The patient began to develop symptoms consistent with worsening catatonia: waxy flexibility, echolalia, muscle rigidity, negativism, stereotypy (lip and tongue biting; fidgeting with clothes), blank staring, and a grasp reflex. She received scheduled doses of lorazepam 2 mg iv every 6 hours, with temporary but dramatic improvement.
The following week, rituximab treatment was attempted, but the full course could not be completed because she developed autonomic instability and worsening catatonia within 24 hours. Emergent electroconvulsant therapy (ECT) was initiated on alternating days, normalizing blood pressure, pulse, and temperature, and improving her cognitive functioning. After seven ECT sessions, she became more aware of her surroundings, could name her location, play a simple card game, eat and toilet with assistance, and ambulate independently. Despite these advances, she remained impulsive and aggressive, and showed sleep-cycle inversion. She was started on valproic acid and trazadone to help stabilize her mood and improve nighttime sleep, while her lorazepam was tapered. The patient showed some improvement in aggression and sleep on this regimen.
To treat her autoimmune disorder, plasma exchange was performed in five sessions, without significant improvement in her cognitive functioning. One month later, rituximab was administered, followed by cyclophosphamide. She was eventually switched over to risperidone 2 mg bid and lorazepam 0.25 mg po bid to help with aggression and anxiety. The patient showed gradual improvement in cognitive functioning after her discharge to an inpatient rehabilitation program. Eight months after her initial symptoms, she began to engage in conversation with her parents, started running track, and could read and write at a fifth-grade level.
Discussion
Over the past 4 years, there has been a rapidly-growing literature on anti-NMDAR encephalitis. After the development of a reliable immunoassay in 2007 by Dalmau and colleagues, over 400 cases have been identified.5,6
Defining Characteristics
This disorder is considered a type of limbic encephalitis (LE) whose etiology was previously unknown. It typically begins with prodromal, nonspecific fever, diarrhea, vomiting, headache, or upper-respiratory symptoms before the onset of psychiatric and neurological sequelae that progress in two stages.1,2
Early-stage symptoms can appear between 1 day and 21 days after prodromal symptoms. A reported 72%–83% of patients present with psychiatric symptoms;1 76%–82% of patients have presented with seizures, which are most often generalized or complex partial seizures.2,3 Early-stage symptoms include amnesia, confusion, memory defects, bizarre behavior, agitation, anxiety, depression, paranoid thoughts, and visual or auditory hallucinations.7 Later-stage symptoms include decreased level of consciousness, lethargy, seizures, hypoventilation, autonomic instability, and dyskinesias (choreoathetoid, orofacial, or limb movements, parkinsonism, rigidity, oculogyric crises, startle, myoclonus, opisthotonus, and movement disorders). Central hypoventilation and autonomic instability often necessitate transfer to an intensive-care unit for ventilatory support, ranging from 2 to 40 weeks.3
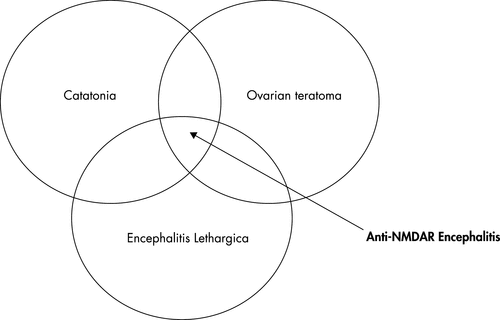
LE can be associated with an underlying paraneoplastic disease, such as an ovarian teratoma, or a non-paraneoplastic process, such as an acquired autoimmune process against neuropil, hippocampal dendrites, or voltage-gated potassium channels (VGKC). Table 1 lists paraneoplastic antibodies associated with limbic encephalitis. Figure 2 demonstrates the anti-NMDAR encephalitis patient population defined by overlapping clinical findings: ovarian teratoma, symptoms of encephalitis lethargica, and catatonia.
| Antibody | Syndrome | Cancer |
|---|---|---|
| Amphiphysin | Limbic encephalitis, stiff-person syndrome | SCLC, breast |
| CV2/CRM, P5 | Limbic, striatal encephalitis (chorea), cerebellar ataxia, peripheral neuropathy, uvitis | Thymoma, SCLC |
| Hu | Limbic encephalitis, encephalomyelitis | SCLC, other |
| Ma Proteins | Limbic, hypothalamic, and brainstem encephalitis | Lung, testis, other |
| NMDA-R | Limbic encephalitis, anti-NMDAR subtype | Ovarian and testicular teratomas, seminoma, SCLC |
Classic LE differs from anti-NMDAR encephalitis in that 70% of LE patients show signal abnormalities on MRI in the medial temporal lobes.8 Half of the patients with anti-NMDAR encephalitis have no abnormalities on MRI.2,6 Occasionally, subtle FLAIR hyperintensities are found in the medial temporal regions.9,10Figure 1 shows our patient’s brain MRI with contrast, showing subtle, nonspecific periventricular hyperintensities on axial FLAIR. No temporal lobe involvement was found.
Temporal-lobe seizures are common in LE and can be demonstrated on EEG as focal epileptic activity in one or both of the temporal lobes or as generalized slow-wave activity.8 Approximately 50% of patients with anti-NMDAR encephalitis have epileptiform discharges, which present early and often evolve into generalized slow waves.2
To further complicate the picture, anti-NMDAR encephalitis has been described as a “dyskinetic encephalitis lethargica” because of overlapping clinical features with the dyskinetic type of encephalitis lethargica.11,12 Encephalitis lethargica (EL) is a subacute or acute encephalitic disorder, with at least three of the following: oculogyric crises, central respiratory irregularity, signs of basal-ganglia involvement, akinetic mutism, somnolence and/or sleep-inversion, and central respiratory irregularities. In a retrospective study of patients age ≤15 years, half of the 20 cases diagnosed with encephalitis lethargica were found to have anti-NMDAR antibodies.11 Parkinsonian symptoms in anti-NMDAR encephalitis occur later than in EL patients without anti-NMDAR antibodies. MRI lesions in EL are more distinctively located in the basal ganglia and substantia nigra/midbrain area.
Anti-NMDAR encephalitis has been recently recognized as a distinct disease, and should be considered in patients who were previously diagnosed with “encephalitis of unclear etiology,” “encephalitis lethargica,” “idiopathic encephalitis,” and “limbic encephalitis.”
Demographics and Tumor Association
Primarily found in younger women, (median age: 22 years), this disorder can affect a wide age-range (23 months to 76 years).2,3,11 A reported 49%–59% of cases have tumors, which are primarily ovarian teratomas.3,4,6 There have been a few cases in young boys and men.2–4 Tumors are identified less frequently in the pediatric population, with the frequency increasing with age; 40% of girls age ≤18 years and 9% of girls ≤14 years have recognizable tumors.4
Some publications have reported anti-NMDAR encephalitis prevalence in predominantly non-Caucasian populations, including Hispanics, African Americans, Asians, and Pacific Islanders.10,13,14 Dalmau et al. found that African American girls are more likely than other ethnic groups to have an underlying ovarian teratoma.6
Symptoms Manifested in Children Versus Adults
There are unique challenges to treatment of anti-NMDAR encephalitis in the pediatric population. Presenting symptoms are harder to recognize in younger children and can manifest as agitation, aggression, temper tantrums, other behavioral changes, and progressive speech deterioration.4 Eventually, stereotyped movements of the face, limbs, and trunk develop, which characterize catatonia. However, these new-onset movements may be misinterpreted as seizures in this younger population. Florance and colleagues recommend a thorough video EEG evaluation before treating cases with antiepileptic medications.4 Also, autonomic dysregulation appears less severe, compared with that in adults. Only 23% of pediatric cases were found to have central hypoventilation, compared with 66% of patients in a primarily adult case series.3,4 Pediatric patients can experience urinary incontinence and insomnia more often than hypersomnia, as seen in our patient.
Pathophysiology
Anti-NMDAR encephalitis appears to be mediated by antibodies to the NR1 subunit of the NMDAR.3 Dalmau et al. has shown in animal models that rats with anti-NMDAR antibodies developed reduced receptor-density in the hippocampus due to capping and internalization of surface NMDARs. It appears that these auto-antibodies cause a selective, yet reversible decrease in NMDARs, which are titer-dependent. The presence of oligoclonal bands, intrathecal synthesis, and pleocytosis in patients with anti-NMDAR antibody-positive CSF suggests that this is an immune disorder.3 Patients with tumors develop higher titers of antibodies, suggesting a stronger immune response than those without tumors.
Dalmau et al. reported that patients experience a persistent amnesia of their convalescence, which was true for our patient.3 This finding suggests that anti-NMDAR antibodies disrupt synaptic plasticity involved in learning and memory. Another study that hints at a mechanism by which these autoantibodies cause behavioral, learning, and memory problems was published by Iizuka and colleagues.15 They found mild frontotemporal atrophy on MRI and hypoperfusion of cerebral blood flow on single photon-emission computed tomography (SPECT) in two cases with severe, protracted cognitive deficits up to 1 year after initial presentation; at 3 and 5 years later, both patients returned to near-baseline cognitive functioning, correlating with significant reversal in frontotemporal atrophy and hypoperfusion on repeat SPECT exam.15
Patients without tumors can recover gradually from cognitive deficits, which correlates with diminishing antibody titers, suggesting that the immune response is not maintained. This brings to question what role a viral-like prodrome plays in the autoimmune sequelae.9 Does a viral infection precipitate or work in conjunction with the teratoma in building an autoimmune response? It has been postulated that a viral infection may trigger an inflammatory response that disrupts the blood–brain barrier, allowing autoantibodies to enter the CNS. This may allow antigen-specific T-cells to aid in intrathecal production of autoantibodies to NMDARs.2 Despite advances in identification of this disorder, the trigger causing the underlying immune response remains unknown.
The NMDAR hypofunction hypothesis of schizophrenia has recently emerged and may help explain psychotic symptoms observed in anti-NMDAR encephalitis.9 Ketamine, phencyclidine, and MK801 are noncompetitive NMDAR antagonists. They selectively inhibit NMDARs on neurons, which release glutamine on GABAergic interneurons. Brown et al. suggest that hallucinations may result from “aberrant activation” of GABAergic interneurons, resulting in delivery of information that is inconsistent in space and time.16 NMDAR antagonists in animal studies cause insomnia and hyperactivity, prolonged changes in non-REM sleep, and hypoventilation.17 In humans, NMDAR antagonists at low doses induce psychotic behavior, memory problems, and decreased response to pain. At higher doses, they can cause dissociative anesthesia, catatonic symptoms, and autonomic disruption, causing hypersalivation, cardiac arrhythmias, and hypertension.3,6 Because of irregular excitatory activity in the limbic, hippocampal, and cortical regions, NMDAR antagonism ultimately leads to unconsciousness and an active slowing EEG pattern.16
Current Treatment
In cases where a teratoma is identified, tumor removal can be curative. The neuropsychiatric symptoms reverse within a few weeks after tumor removal with the addition of IVIG or iv steroids.10 Immune suppression is the primary method for addressing the underlying autoimmune process, regardless of whether a tumor has been identified. Treatment should begin with iv corticosteroids, and should also include plasma exchange or IVIG.6 If the patient has an inadequate response, deteriorates, or relapses, treatment with rituximab, cyclophosphamide, or both is indicated. Chronic immune suppression with mycophenolate mofetil or azathioprine has been proposed.6 Better rates of recovery have been observed with fewer relapses in patients who receive early immunotherapy.3
Recovery is a gradual process, in part due to the limitations of current treatment options to maintain control of the CNS immune response. While serum titers diminish, CSF titers decline at a slower rate.3 Those patients with tumors tend to have higher antibody titers than those without tumors. Milder symptoms also tend to correlate with lower titers. As described in a few reports, some patients who have shown a slow, protracted recovery have been found to have ovarian teratomas on MRI years after initial onset of symptoms.15 Although it is possible for patients without tumors to recover completely, they tend to recover much more gradually. MRI and PET scan may not be sensitive enough to pick up slow-growing microtumors, thus requiring several years before detection. Patients with no identifiable tumor should undergo yearly screening by pelvic MRI.4,6
For treatment of psychiatric symptoms—mood lability, aggression, impulsivity, and hallucinations—only one publication focuses on pharmacologic management.18 Typical and atypical antipsychotics have been used with limited success for aggression and hallucinations. In extreme cases of agitation, phenobarbital and fentanyl have been used to induce a medical coma. Chapman et al. report improvement in sleep-cycle regulation with clonidine, trazodone, and benzodiazepenes. Insomnia was treated with some success in our patient using diphenhydramine. For mood dysregulation, valproic acid and lithium have shown only a minor effect, as was observed in our patient after a trial of valproic acid. For hyperactivity and impulsivity, a 6-year-old boy was responsive to psychostimulants.4
Antipsychotics, such as haloperidol, must be used with caution because they can exacerbate movement disorders in the later stages of the disease-progression, when patients can develop catatonia.4,18 Neuroleptics may confuse an already-complex clinical picture because several patients treated with haloperidol have later developed autonomic instability, elevated CK, and rigidity, which is characteristic of both neuroleptic malignant syndrome and malignant catatonia.6 A test dose of lorazepam (1 mg–2 mg) can be used to confirm the diagnosis of catatonia.
Catatonic features can be ascribed to multiple psychiatric disorders, and require identification of two or more criteria (Table 2).19,23 The DSM-IV describes catatonia as a qualifier and subtype of mood or psychotic disorders. In the development of DSM-V, catatonia as a separate entity has been debated.
| Motor immobility as evidenced by catalepsy (including waxy flexibility) or stupor |
| Excessive motor activity (purposeless, not influenced by external stimuli) |
| Extreme negativism (motiveless resistance to all instructions or maintenance of a rigid posture against attempts to be moved) or mutism |
| Peculiarities of voluntary movement as evidenced by posturing, stereotyped movements, prominent mannerisms, or prominent grimacing |
| Echolalia or echopraxia |
Treatment of catatonia requires benzodiazepines at regular intervals, as seen in our patient who was given lorazepam 2 mg iv q 6 hrs. Some cases may require up to 20 mg–30 mg lorazepam daily to alleviate symptoms.19-22 ECT is the gold standard when patients fail to respond to increasing doses of benzodiazepines, or show signs of malignant catatonia.21,22 Malignant catatonia is a severe, lethal form of catatonia, in which patients exhibit abrupt muteness, unresponsiveness, echopraxia, echolalia, psychomotor changes, autonomic instability, and fever.22
There are few published reports on the use of electroconvulsive therapy (ECT) in adolescents, and fewer in patients with catatonia secondary to anti-NMDAR encephalitis.4,20,21 Although the number of treatments required varies greatly, patients with catatonia require more frequent ECT than those being treated for refractive depression. In one case report, seven bilateral ECT treatments were required in an anti-NMDAR encephalitis case with catatonia before sustained clinical improvement was achieved. The death rate in patients with untreated malignant catatonia is 10%–20%. The efficacy rates for treatment of catatonia are estimated to be 80%–100% with lorazepam, and 82%–96% with ECT.19 In the pediatric population (age 18 years and younger), ECT was found to be effective in 80% of cases of catatonia.24 The adverse effects of ECT in young people are similar to those described in adults, with headache as the most frequent complaint. Muscle soreness, nausea, acute confusion, and time-limited anterograde and retrograde memory loss are also common.22
ECT is rarely used in the pediatric population because of stigma and limited number of patients presenting with psychiatric illness severe enough to warrant petitioning its use. Even when parents consent, there are difficulties obtaining legal permission for its use in certain states. Although the exact mechanism of action remains unclear, animal models have shown that ECT causes up-regulation of NMDA-receptors, which hints at how it has been beneficial in patients with anti-NMDA encephalitis.25 It may be an important intervention for treatment of the underlying disease process.
Prognosis
All patients require help from a multispecialty team, which includes physical therapists and rehabilitation specialists. Relapse or incomplete recovery is more common in patients without identifiable tumors or with relapsing teratomas.4 In a case series consisting primarily of adults, 75% of patients recovered their premorbid cognitive functioning, whereas 25% had persistent, severe deficits or died.3 Relapse has been reported in approximately 15%–25% of cases.6,11 In a study of children and adolescents, 9 of 31 patients had full recovery; 14 of 31 had substantial improvement, but with mild deficits; and 8 of 31 had limited improvement, with signs of severe deficits.4 Gable et al. report that 10% of NMDAR-encephalitis cases, ranging in age from 11 years to 31 years, resulted in death.
Conclusion
Psychiatrists should have a high suspicion for anti-NMDAR encephalitis in young, female patients with acute seizures, associated with abrupt behavioral changes or dyskinesias. These symptoms, coupled with evidence of CSF lymphocytic pleocytosis or oligoclonal bands, an EEG with generalized slow-wave activity, and nonspecific, hyperintensities on brain MRI help support clinical suspicions. Full recovery is obtained more often in patients who are started early on immune-suppressant therapy. Benzodiazepines and ECT may be warranted for treatment of progressive, life-threatening autonomic instability and muscle rigidity associated with malignant catatonia.
Additional research is necessary to determine the optimal treatment for this disease and its psychiatric manifestations. The best outcomes result from cooperation within a multidisciplinary team to address the needs of such medically complex patients.
1 : Anti-NMDA-receptor encephalitis: a severe, multistage, treatable disorder presenting with psychosis. J Neuroimmunol 2011; 231:86–91Crossref, Medline, Google Scholar
2 : N-methyl-D-aspartate antibody encephalitis: temporal progression of clinical and paraclinical observations in a predominantly non-paraneoplastic disorder of both sexes. Brain 2010; 133:1655–1667Crossref, Medline, Google Scholar
3 : Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol 2008; 7:1091–1098Crossref, Medline, Google Scholar
4 : Anti-N-methyl-D-aspartate receptor (NMDAR) encephalitis in children and adolescents. Ann Neurol 2009; 66:11–18Crossref, Medline, Google Scholar
5 : Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol 2007; 61:25–36Crossref, Medline, Google Scholar
6 : Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol 2011; 10:63–74Crossref, Medline, Google Scholar
7 : Limbic encephalitis presenting with seizures, anterograde amnesia, and psychosis in a patient seven weeks status post-immature ovarian teratoma removal. Mil Med 2010; 175:616–618Crossref, Medline, Google Scholar
8 : Clinical and immunological diversity of limbic encephalitis: a model for paraneoplastic neurologic disorders. Hematol Oncol Clin North Am 2006; 20:1319–1335Crossref, Medline, Google Scholar
9 : Anti-NMDA receptor encephalitis in Japan: long-term outcome without tumor removal. Neurology 2008; 70:504–511Crossref, Medline, Google Scholar
10 : Anti-N-methyl-D-aspartate receptor encephalitis associated with an ovarian teratoma in an adolescent female. J Pediatr Surg 2010; 45:1550–1553Crossref, Medline, Google Scholar
11 : N-methyl-D-aspartate receptor antibodies in pediatric dyskinetic encephalitis lethargica. Ann Neurol 2009; 66:704–709Crossref, Medline, Google Scholar
12 : A modern perspective on the differential diagnosis between encephalitis lethargica or anti-NMDA-receptor encephalitis. J Clin Neurosci 2010; 17:1204–1206Crossref, Medline, Google Scholar
13 : Anti-NMDA receptor encephalitis: report of ten cases and comparison with viral encephalitis. Eur J Clin Microbiol Infect Dis 2009; 28:1421–1429Crossref, Medline, Google Scholar
14 : Acute psychiatric illness in a young woman: an unusual form of encephalitis. Med J Aust 2009; 191:284–286Crossref, Medline, Google Scholar
15 : Reversible brain atrophy in anti-NMDA receptor encephalitis: a long-term observational study. J Neurol 2010; 257:1686–1691Crossref, Medline, Google Scholar
16 : General anesthesia, sleep, and coma. N Engl J Med 2010; 363:2638–2650Crossref, Medline, Google Scholar
17 : Severe childhood encephalopathy with dyskinesia and prolonged cognitive disturbances: evidence for anti-N-methyl-D-aspartate receptor encephalitis. Dev Med Child Neurol 2010; 52:e78–e82Crossref, Medline, Google Scholar
18 : Anti-NMDA receptor encephalitis: diagnosis, psychiatric presentation, and treatment. Am J Psychiatry 2011; 168:245–251Crossref, Medline, Google Scholar
19 : The catatonia syndrome: forgotten but not gone. Arch Gen Psychiatry 2009; 66:1173–1177Crossref, Medline, Google Scholar
20 : Pearls and oysters: electroconvulsive therapy in anti-NMDA receptor encephalitis. Neurology 2010; 75:e44–e46Crossref, Medline, Google Scholar
21 : Electroconvulsive therapy in a pediatric patient with malignant catatonia and paraneoplastic limbic encephalitis. J ECT 2006; 22:267–270Crossref, Medline, Google Scholar
22 : Catatonia is hidden in plain sight among different pediatric disorders: a review article. Pediatr Neurol 2010; 43:307–315Crossref, Medline, Google Scholar
23 : Diagnostic criteria from DSM-IV-TR. Washington, D.C., American Psychiatric Association, 2000Google Scholar
24 : Half a century of ECT use in young people. Am J Psychiatry 1997; 154:595–602Crossref, Medline, Google Scholar
25 : Differential effects of electroconvulsive shock on the glutamate receptor mRNAs for NR2A, NR2B, and mGluR5b. Brain Res Mol Brain Res 1998; 61:108–113Crossref, Medline, Google Scholar

