
Case Reports of Rasmussen’s Syndrome and Literature Review
Abstract
Rasmussen’s encephalitis (RE) is a chronic inflammatory disease of unknown origin that usually affects one hemisphere of the brain. Serial electroencephalographic (EEG) and magnetic resonance imaging (MRI) associated with magnetic resonance angiography (MRA) features specific for RE are rarely reported. By assessing the progression of RE revealed through EEG, MRI/MRA, and treatment results, the authors determined that RE is a focal epileptic syndrome characterized by chronic and progressive partial epileptic seizures, with specific changes in EEG and MRI/MRA findings.
Rasmussen’s encephalitis (RE) is a chronic inflammatory condition of unknown etiology that occurs mainly in childhood. This disorder usually affects one hemisphere of the brain and is characterized by intractable epilepsy and progressive deterioration of mental and neurological functions.1,2 Magnetic resonance imaging (MRI) shows a continuous spread of signal alterations in one brain hemisphere, and the change is progressive.3 A diagnosis is usually based on clinical and neuroradiological features, as well as histopathological evidence of inflammatory infiltrates throughout the affected hemisphere.4 The progressive changes of this disorder are rarely detected using serial electroencephalography (EEG), MRI, and magnetic resonance angiography (MRA) together.
We herein reviewed two cases of RE in which clinical, EEG, and MRI associated with MRA features of disease progression were identified and used to make an early diagnosis and develop an effective treatment plan.
Case Reports
Case 1
A boy, age 2 years and 8 months, presented with seizures characterized by sudden onset of unstable gait, accompanied by left limb hypodynamia in the morning. Several minutes after each seizure, the child experienced a unilateral oral twitch and deviation of head and eyes, which were associated with a secondary generalized seizure. The seizures, which occurred several times daily, decreased in severity after an intramuscular injection of phenobarbitone (PB) and were seen only intermittently (once every few days) thereafter.
Two months after PB treatment, the patient fell suddenly, exhibiting hemiconvulsion, posthemiparesis, and a positive Babinsiki’s sign on the left side of his body. Various symptoms of seizures were then seen, including oral twitching and staring, as well as signs of hemiclonic seizure. On several occasions, the patient demonstrated unilateral status epilepticus (SE), gradually progressing to epilepsia partialis continua (EPC), which was gradually transformed into intermittent focal clonus. He then responded to intravenous midazolam. Since discharge from the hospital, he has complained of repeated somatosensory symptoms, and experienced a focal seizure and drop attack on the left side of his body, despite continuous oral administration of carbamazepine (CBZ) 300 mg/day and prednisolone (PDS) 20 mg/day.
By Month 11 after the onset of illness, the patient still exhibited focal seizures, along with unilateral neurological and cognitive deficits, although not as severe as before. After topiramate 75 mg/day po was added, a striking improvement in seizure control and cognition was observed. However, the patient still experienced hemiparesis and occasional drop attacks.
During the first onset of illness, immunological tests of his serum were positive for cytomegalovirus (CMV) immunoglobulin (Ig) (CMV-IgG). Chemistry and virology tests of the patient’s cerebrospinal fluid were normal, but IgM (0.069 mg/dL) was markedly higher than normal (0.0011–0.0370 mg/dL).
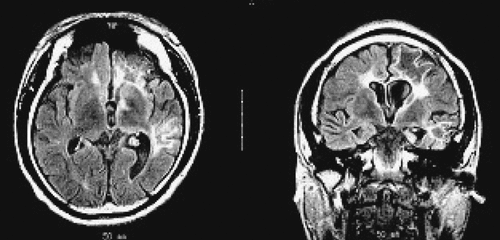
The EEG taken during the initial stage of disease revealed diffuse, high-amplitude, polymorphic delta activity over the affected hemisphere, especially over the right frontal lobe. This activity shifted to the left frontal region for a short time (1 month) and became a distinctive early feature of his condition (Figure 1 [A–C]). After 9 months, EEG revealed progressive impoverishment of background activity over the affected hemisphere and dominant spike-wave activity over the right frontotemporal area during sleep (Figure 1 [D, E]). His initial MRI revealed an obvious focal area consisting predominantly of edema coinciding with a depression in the ipsilateral ventricle. The hemispheric ratio (HR) was 1.26, which indicates the earliest stage of RE. Within 9 months, HR declined to 0.72, indicating a considerable amount of atrophy, and enlargement of the lateral ventricle on the involved side of brain (Figure 2 [A, C])). The MRA revealed a distinctive feature of a later stage of the disease: a mild depression and shift in the location of the cerebral middle artery (CMA), progressing to a later stage characterized by the absence of the CMA and cerebral anterior artery (CAA; Figure 2 [B, D]).
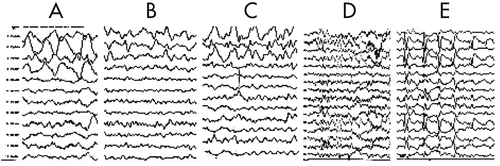
[A] Right-frontal predominantly diffuse delta activity of high amplitude during comatose period; [B] Relatively focal delta activity in the right-frontal area of hemisphere 1 month after seizure were controlled; [C] Left-frontal predominance of delta activity during sleep at the same time as B; [D] Right-frontal dominance of diffuse slowing background activity 9 months after seizures were controlled; [E] Epileptic abnormality mainly involving the right fronto- temporal regions.
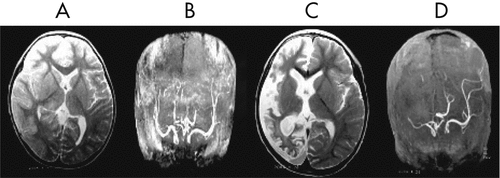
[A] MRI showed a diffuse signal hyperintensity over the affected region of bilateral hemispheres, mainly involving the right fronto-temporal and the caudate head, putamen and thalamus, coincided with the ipsilateral ventricle depression; [B] MRA indicated that right CMA was slightly depressed; [C] Nine months later, MRI showed a noticeable cortical atrophy of the right hemisphere, with ipsilateral ventricle dilatation and midline line-shifting to the right hemisphere; [D] Deficit of right CMA and CAA caused by inflammatory ventricle lesion.
Case 2
A boy 6 years and 4 months old experienced a head injury during a fall. He then experienced a hemiconvulsion on the right side of his body that continued for about 10–20 minutes, accompanied by ipsilateral eye deviation, dysarthria, and transient postictal hemiparesis. The seizure recurred about 1–2 times per month, despite treatment with PB 90 mg/day. Five months later, the disorder became aggravated, characterized by frequent asymmetric clonic seizures, accompanied by eye deviation, speech deficit, and oral and manual automatism. These symptoms progressed to unilateral or generalized motor manifestations. During this period, he demonstrated a transient response to CBZ 500 mg/day and PB 90 mg/day. At age 6 years and 11 months, the patient still experienced illness progression, which was characterized by several types of seizures, including simple partial seizures, occasional EPC, or prolonged ipsilateral complex partial seizures or secondary generalized tonic–clonic seizure (sGTCS), associated with mental and neurological deterioration.
Neither CBZ 600 mg/day nor valproate (VPA) 800 mg/day were useful for this patient, so human intravenous immunoglobulin (IVIG) 400 mg/kg/day was given by infusion for 5 days. Seizure frequency decreased, and neurological function improved after 3 additional infusions of IVIG monthly. Because of intermittent partial seizures (PS), EPC, and sGTCS, PDS 40 mg/day was administered, along with oxcarbazepine (OXC) 600 mg/day and PB 90 mg/day. At the 1-year follow-up visit, seizures were markedly controlled, and mental and neurological functions were greatly improved. The patient still experienced PS and occasional drop attacks once daily or every several days, but refused surgery because of the risk of damage to speech functions.
An EEG revealed that a diffuse slowing background interposed with focal rhythmic epileptic activity had spread over the entire brain in association with sGTCS (an early feature of RE; Figure 3). Five months later, EEG showed diffuse reduced activity, intermingled with epileptic abnormalities over the left frontal-temporal-parietal regions that coincided with PS, EPC, dysarthria, and automatism, and served as another distinctive feature of the disease (Figure 4 [A–C]). The MRI revealed diffuse cerebral atrophy intermingled with focal atrophy accompanied by dilation of the left lateral ventricle of the brain (Figure 5).
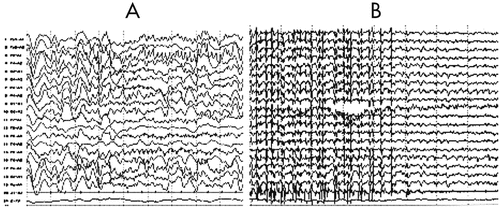
[A] Ictal EEG showed the rhythmic sharp-waves of the left frontotemopral region superimposed on the diffuse slowing background activity; [B] the left, predominantly diffuse, spike-wave discharges followed by diffuse slowing background activity.
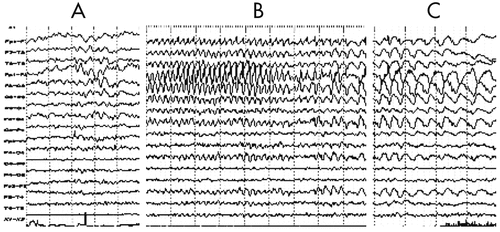
[A] Interictally, focal theta and delta slowing activities predominantly exhibited on the left fronto-temporal-central regions; [B] Ictally, focal rhythmic theta activity of high voltage, with interposed small, sharp waves were dominantly superimposed on the diffuse rhythmic slowing background. Clinically, oral and manual automatism, and tonic seizures of the right limb were noted; [C] Postictally, focal delta activity of more slowing and higher voltage in the same region as above was accompanied by gradual remission of right-gesture seizures.
Literature Review and Discussion
These two cases demonstrate the sudden onset of RE presenting as unstable gait or after a head injury. Each incident was followed by partially dominant seizures that developed into sGTCS during the prodromal phase, when seizures are not frequent and are accompanied by transient or mild hemiparesis. These seizures occurred over a period of approximately 2 to 5 months, which is shorter than the median of 7.1 months reported for patients with Type 1 disease5 and that might have been related to a high incidence of infectious disease and the resulting abnormal autoimmunization in children. Frequent seizures, which define the acute phase of the disease, occurred over 5 to 9 months, in our cases, which is slightly longer than the median duration of 4 to 8 months reported for Type 1 patients.4 Our experience indicates that an early diagnosis, effective patient cooperation, and appropriate treatment are crucial for a good prognosis for patients with RE.
Bien3,6 divided RE into three phases: early, acute, and residual. The early (prodromal) phase is characterized by less-frequent seizures, occurring over a period of several weeks to months. During this phase, patients do not have obvious abnormal EEG or MRI findings, which makes it difficult to make an early diagnosis. For this reason, additional clinical evidence, for example, repetitive PS or sGTCS plus slight or transient hemiparesis, should be taken into consideration when making a diagnosis. In Case 1, the patient exhibited infrequent PS and sGTCS associated with focal edema and diffuse rhythmic slowing activity. The acute (progressive) phase is characterized by frequent focal motor seizures and sGTCS or EPC with hemiparesis and intellectual deficits lasting several months to several years or longer. In our experience, an early diagnosis and appropriate treatment during this phase are effective in reducing the symptoms of this illness and improving the prognosis. The residual phase is characterized by a decreased frequency or cessation of seizures accompanied by mental and neurological deficits. In both of the cases presented in this study, this third phase developed after 10–11 months of illness accompanied by intermittent and various types of PS.
The etiology of RE is unknown, although some clinical and experimental findings support a role for certain viral infections or autoimmunity involving the glutamate receptor (GluR).2,3,7 In most patients, the trigger for the abnormal immune response is unknown, although RE is known to follow an infection or head injury.6,8 The detection of CMV-IgM/IgG in patient’s serum and a mixture of predominantly diffuse and somewhat focal cytotoxic edema (on MRI) suggest a role for a strong inflammatory episode (possibly due to a viral infection and the autoimmune response that follows), followed by the destruction of local tissue.4,9,10 The history of a head injury in Case 2 implicates that exposure of GluR3 caused by focal breakdown of the blood–brain barrier can trigger an autoimmune reaction.9,11
The acute-phase EEG taken in Case 1 showed that the diffuse slowing activity occurred primarily in the right hemisphere, but shifted between the two hemispheres for a short period. It also revealed cytotoxic edema in a white-matter lesion that arose as a result of an inflammatory episode during SE. Nine months later, EEG showed diffuse slowing activity, intermingling with focal epileptiform discharges in the right anterior region of the brain. This is an unusual EEG presentation and might be complementary evidence that can be taken into consideration to make an early diagnosis. In Case 2, the seizure correlated with focal rhythmic slowing waves and interposed aborted spikes in the left frontaltemporal region 12 months after the patient was first seen. The progression of generalized slowing background activity toward focal (primarily slow) waves or toward spike-wave discharges was a distinctive feature in both cases. These patterns have not been uncommonly reported in the literature.12,13
High signal-intensity on T2-weighted MRIs taken during the initial stage of the disorder in Case 1 revealed unilateral cortical swelling, followed by progressive atrophy and destruction of the CAA and CMAs in the affected hemisphere. In Case 2, the MRI revealed areas dominated by diffuse brain atrophy in some focal areas—another typical feature of RE. Fiorella and Kim14,15 identified two distinct patterns of atrophy in RE: 1) a uniform diffuse pattern involving mainly the cerebral hemisphere; and 2) a pattern of focal atrophy superimposed on a background of diffuse atrophy. The HR, that allows us to compare the extent of change in one hemisphere with the extent of change in the other, consisted of the ratio of pixels in the affected versus the unaffected hemisphere. An HR value of 1.0 indicates that both hemispheres on the assessed slices were equal in size; a value >1.0 indicates that the affected hemisphere is larger; and a value or <1.0 indicates the affected hemisphere has undergone a greater degree of atrophy than the unaffected one. Thus, the HR can be used for an early-stage diagnosis of RE. In Case 1, HR was 1.26 during the initial phase, indicating that swelling had occurred primarily in the affected hemisphere. It declined to 0.72 during the period between the acute and residual phases, which strongly suggests a remarkable degree of atrophy in the affected hemisphere.4
RE strongly resembles hemiconvulsions-hemiplegia-epilepsy syndrome (HHES), which can be identified by the presence of prolonged complex febrile seizures or the onset of febrile status convulsions before age 4 years.16 Immediately after its initial onset, HHES is characterized by a flaccid hemiplegia that varies in duration; this is followed by a PS that usually originates in the temporal lobe. Other diseases characterized by SE, especially when it is associated with cortical dysplasia, should be ruled out on the basis of these clinical signs.
1 : Focal seizures due to chronic localized encephalitis. Neurology 1958; 8:435–445Crossref, Medline, Google Scholar
2 : Imaging of chronic encephalitis, in Chronic encephalitis and epilepsy: Rasmussen’s syndrome. Edited by Andermann F. Boston, Butterworth- Heinemann, 1991, pp 47–60Google Scholar
3 Laurent Sheybani, Karl Schaller, Margitta Seeck. Rasmussen’s encephalitis: an update. Schweizer archivfur neurologienund psychiatrie 2011;162(6):225–31.Google Scholar
4 : Destruction of neurons by cytotoxic T-cells: a new pathogenic mechanism in Rasmussen’s encephalitis. Ann Neurol 2002a; 51:311–318Crossref, Medline, Google Scholar
5 : Diagnosis and staging of Rasmussen’s encephalitis by serial MRI and histopathology. Neurology 2002b; 58:250–257Crossref, Medline, Google Scholar
6 : Rasmussen’s encephalitis with concomitant cortical dysplasia: the role of GluR3. Epilepsia 1999; 40:242–247Crossref, Medline, Google Scholar
7 : Autoantibodies to glutamate receptor GluR3 in Rasmussen’s encephalitis. Science 1994; 265:648–651Crossref, Medline, Google Scholar
8 : Autoantibodies to NMDA receptor in patients with chronic forms of epilepsia partialis continua. Neurology 2003; 61:891–896Crossref, Medline, Google Scholar
9 : Rasmussen’s Syndrome: Epileptic Syndrome in Infancy, Childhood, and Adolescence, 3rd Edition. John Libbey & Ltd, 2002, chap 32, 495–511Google Scholar
10 : Limited chronifocal encephalitis: another variant of Rasmussen syndrome? Neurology 2008; 70:374–377Crossref, Medline, Google Scholar
11 : A substantial number of Rasmussen syndrome patients have increased IgG, CD4+ T cells, TNFalpha, and Granzyme B in CSF. Epilepsia 2009; 50:1419–1431Crossref, Medline, Google Scholar
12 : Clinical and electroencephalographic correlates in Rasmussen’s encephalitis. Epilepsia 1997; 38:189–194Crossref, Medline, Google Scholar
13 : Rasmussen’s encephalitis: early characteristics allow diagnosis. Neurology 2003; 60:422–425Crossref, Medline, Google Scholar
14 : (18)F-fluorodeoxyglucose positron emission tomography and MR imaging findings in Rasmussen encephalitis. AJNR Am J Neuroradiol 2001; 22:1291–1299Medline, Google Scholar
15 : A longitudinal MRI study in children with Rasmussen syndrome. Pediatr Neurol 2002; 27:282–288Crossref, Medline, Google Scholar
16 : The HHE Syndrome. Epileptic Syndrome in Infancy, Childhood, and Adolescence, 4th Edition. John Libbey & Ltd, 2005, chap 17, 277–293Google Scholar

