
The Neuropsychiatry and Multisystem Features of the Smith-Magenis Syndrome: A Review
Abstract
Smith-Magenis Syndrome (SMS) is a complex, pediatric, neurobehavioral, contiguous gene syndrome ascribed to interstitial microdeletion of chromosome 17, band 11.2. The syndrome is characterized by distinctive behavioral, neurocognitive, and neuropsychiatric abnormalities. This genetically mediated disorder of mental retardation prompts behavioral researchers to examine the links between genes, brain, and behavior in order to solve the gene-behavior puzzle and the genotype/phenotype correlation. In this article, the authors review literature on behavioral profile and its associated psychopathologies, cognitive profiles, multisystem abnormalities, and genetic correlates that highlight the complexities of the disorder.
Smith-Magenis Syndrome (SMS) is a complex neuropediatric-neurobehavioral syndrome associated with an interstitial microdeletion aberration at chromosome 17p11.2. It was in 1982 when the first original description was reported by the genetic counselor Ann C. M. Smith and her colleagues, including geneticist Ellen Magenis.1 Since then, more than 100 cases have been described in the literature.
Smith-Magenis syndrome is a distinctly characterized genetically driven disorder exemplified by multiple congenital anomalies and mental retardation, distinct craniofacial dysmorphic phenotype, abnormalities of sleep-wake circadian rhythm, severe cognitive and behavioral profiles, and other psychopathologies. With the advent of powerful cytogenetic and molecular laboratory tools, it has been possible to recognize the distinctive genetic underpinnings of various neurobehavioral disorders.
In 1986, Smith et al.2 described nine individuals with chromosomal deletion at 17p11.2, which was subsequently confirmed in 27 SMS patients by the multidisciplinary clinical and cytogenetic study of Greenberg et al.3 in 1991. The birth prevalence of SMS is believed to be as high as 1 in 25,000. The clinical phenotype has been well described and includes minor craniofacial anomalies such as: brachycephaly; a prominent forehead; a flat midface; a down-turned mouth with cupid’s bow; synophrys; epicanthal folds; a broad nasal bridge; atypical ears; and prognathism. Other abnormalities include a hoarse, deep voice; a short stature; broad hands with brachydactyly; and self-injurious behaviors such as head banging, wrist-biting, onychotillomania, and polyembolokoilamania.3 Sleep disturbances; self-hugging (auto-amplexation) stereotypy; signs of peripheral neuropathy (decreased deep tendon reflexes, decreased sensitivity to pain, bony markers of pes cavus or planus); delayed speech and motor development; and variable levels of mental subnormalities are also common.
The seminal multidisciplinary studies of Greenberg et al.4 conducted on 27 SMS patients demonstrated the otolaryngologic abnormalities in 94%, eye abnormalities in 85%, rapid eye movement (REM) sleep abnormalities in 75%, hearing impairment in 68% (65% conductive and 35% sensorineural); cardiac abnormalities in 37%; scoliosis in 65%; brain abnormalities such as ventriculomegaly in 52%; renal anomalies in 35%; low thyroxine levels in 29%; low immunoglobulin levels in 23%; forearm abnormalities in 16%; and other minor abnormalities such as seizures and genital abnormalities. Mental retardation was scored as falling in the moderate range, varying between an intelligence quotient (IQ) of 40 and 54. It is evident from the frequency of these many abnormalities in SMS that the patients needed mandatory periodic thorough evaluations both at the time of diagnosis and at least on an annual basis thereafter.
Neuropsychiatry and Behavioral Profile
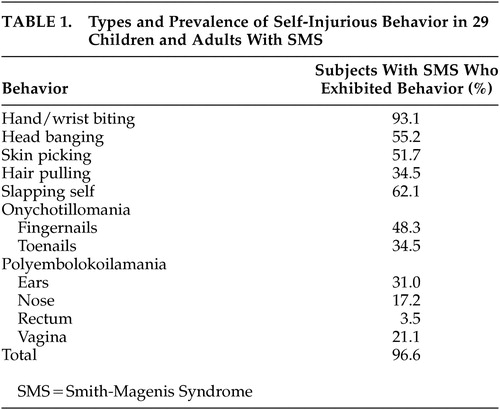
Several investigators5–10 have further elucidated the peculiar clinical and peculiar behavioral phenotype of SMS patients. A characteristic pattern of stereotypic and self-injurious behavior distinguishes SMS from many of the other genetic syndromes of mental retardation.11 In their study of 29 children and adults with SMS, Finucane et al. 12 examined 11 specific types of self-injurious behavior from the prevalence and severity (self-injurious behavior severity ratings) perspective (Table 1). Self-injurious behavior was almost universally present (96.6%) in these patients and may be labeled as “hallmarks” of the SMS. These “genetically driven” behaviors, in addition to their erratic sleep patterns and maladaptive behavior, pose severe management problems and inflict a very stressful burden on the parents who want to protect them from harm. It was also suggested that, with increasing age and ability levels, people with SMS add to their repertoire of self-injurious behavior (self-pinching/scratching, picking skin around the fingernails), which may be due to an operant control of behavior. Alternatively, this could reflect age-dependent gene expression.
The behavioral findings of maladaptive vulnerabilities included attention seeking, hostility, impulsiveness, temper tantrums, aggression, anxiety, sleep problems, overactivity, destruction, overdependency, and a curious set of unusual motor behaviors. Finucane et al.12 have described this unusual motor behavioral phenotype in 11 SMS patients. These included self-hugging behaviors: crossing both arms tightly across the chest; pulling the chin downward and spasmodically tensing the upper body for a few seconds at a time; clasping the hands at chest level or under the chin with fingers interlocked, while squeezing the arms tightly against the sides. These movements would last for a few seconds, and patients would experience sudden “bouts” of several spasms per episode. Finucane et al. described the “spasmodic upper-body squeeze” (self-hug) as “tic-like,” and it was exacerbated by happiness, excitement, or overstimulation, accompanied by facial grimacing and occasional grunting. This behavior is a major clinical clue to the diagnosis, which should prompt chromosomal analysis. Most affected individuals are affectionate and have a positive, affective mood that can be regarded as one of the more benign and appealing aspects of their behavioral phenotype. Among children with SMS, there have been single cases reported on those who fulfill the diagnostic criteria for autism.2,13 Many cases have been reported on those with autistic-type behaviors.14
Cognitive Profile
The behavioral and cognitive functioning profiles were systematically studied in 10 patients with SMS, using various neurobehavioral and neuropsychological assessments conducted by Dykens et al.15 The cognitive abnormalities described included problems in sequential processing (Kaufman Assessment Battery for children) and problems in learning and retention. It was uncertain whether these deficits were attributed to the associated inattention, overactivity and impulsivity.
To our knowledge, the largest cohort of SMS patients studied was comprised of 29 children and 21 adults and was examined in relation to intellectual, cognitive, adaptive behavior, and learning domains by Udwin et al.16 All patients had mild to severe learning disabilities, and levels of attainment and of adaptive behavior were strikingly low. Children with an IQ of 50 or below constituted 75%. In contrast, the adult sample obtained higher IQs but they were more dependent on caretakers, with little independence in daily living skills. Occupational attainment was also low, which was explained on the basis of the high rates of severe behavioral disturbances (aggression and self-injury), poor concentration, impulsivity, attention seeking behaviors, and autistic behaviors that are the typical comorbid psychopathologies of this syndrome.
Dysmorphic Craniofacial Profile
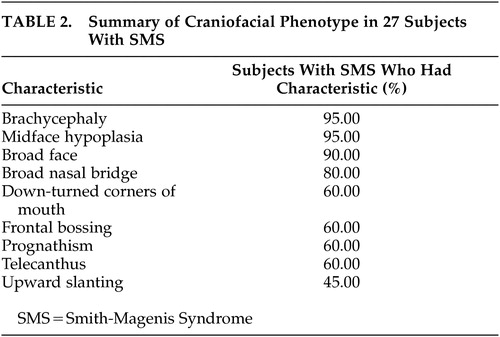
Allanson et al.17 studied the craniofacial patterns of SMS using various facial anthropometric measurements and facial “gestalt” and noted the distinctive facial phenotype. The summary of the craniofacial pattern abnormalities is shown as Table 2. It was also emphasized that these characteristic facial dysmorphic phenotypic features evolve over time and are more subtle in early childhood. Age also enhances the somewhat coarse appearance of the face with development of a hoarse, deep voice and causes an increased mandibular growth that exceeds that of the maxilla, which leads to increased jaw width and protrusion and marked midface hypoplasia.
Circadian Sleep Disturbances and Behavior
Sleep pattern disturbances are common in children with mental retardation and neurodevelopmental disabilities.18 The specific sleep disturbances in SMS include disrupted sleep-wake cycle, early sleep onset, frequent awakenings, early sleep offset, advanced sleep phase and defects in REM sleep. These sleep disturbances have a major impact on patients with SMS and their families. It has been hypothesized that the behavioral problems, hyperactivity, and attention deficits could be related to sleep deprivation and inappropriate diurnal melatonin release in SMS. The use of melatonin at the end of the day has been recently suggested for the treatment of chronic sleep disorders in neurologically disabled children.
DeLeersnyder et al.19 studied another major problem of severe sleep abnormalities in 20 children with SMS and suggested that the behavioral abnormalities could be aggravated by inversion of the circadian rhythm of melatonin secretion. Potocki et al.20 had earlier reported alterations in circadian rhythms causing severe sleep disturbance and advanced sleep phase in SMS by observing the abnormal rhythm of 6 sulfatoxymelatonin, the main urinary melatonin metabolite. DeLeersnyder et al.19 studied the circadian variation of plasma melatonin in children and a comparison group and correlated tantrums with melatonin rise. They hypothesized that at least part of the hyperactivity and attention deficit and behavioral problems occurred because the patients struggled against sleep, when the melatonin rose during the day. Since a number of clock genes control circadian patterns in the suprachiasmatic nuclei, a haploinsufficiency for a clock gene; an alteration of the output/input signaling pathway or an abnormal transmission from suprachiasmatic nuclei to an output signaling pathway of postganglionic fibers ascending to pineal gland could account for the sleep disturbances in SMS.
DeLeersnyder et al.21 examined the effects of beta-1 adrenergic blockers (acebutolol 10 mg/kg as single morning dose) in nine SMS children. They noted a significant improvement of inappropriate behavior with increased concentration, delayed sleep onset, increased hours of sleep, and delayed waking. Melatonin administration is not warranted because the amount of hormone is largely normal but its kinetics are erratic. The study showed that a single morning dose of acebutolol was well tolerated and suppressed the inappropriate diurnal secretion of melatonin which helped to manage hyperactivity, enhance cognitive performance, and reduce sleep disorders in SMS. The abnormal circadian rhythm of melatonin in SMS and its relationship with chromosome 17 deletion remain unclear.
Ongoing trials are underway using various beta-adrenergic antagonists, with different half life kinetics, combined with evening melatonin administration to restore the physiological circadian rhythm of melatonin in SMS.
Multisystem Involvement
Several investigators4,22–26 have reported various features of multisystem involvement in SMS. These clinical findings are summarized as Table 3. It is crucial that every SMS patient should have annual evaluations for thyroid function, scoliosis, and ocular problems. Newly diagnosed patients should be screened for the various anomalies by renal ultrasonography, audiologic evaluation, echocardiogram, assessment for velopharyngeal incompetence, and quantitative immunoglobulins. This would facilitate further appropriate clinical evaluation and early intervention.
Neurological Involvement
Other neurological abnormalities reported in SMS patients include peripheral neuropathy with wasting of the distal musculature with delayed nerve conduction velocities and nerve biopsy showing segmental demyelination and remyelination.2,27 Clinical signs of peripheral neuropathy have been found in approximately 75% of SMS patients.4 Seizures (30%) have been reported, but encephalogram (EEG) epileptiform abnormalities without a clinical history of seizures (21%) have also been described.4 Several adults aged in their 60s and 70s have been described in literature,4 suggesting that life expectancy may be normal.
Genetic Implications—Cytogenetic and Molecular Studies
At a period of growing excitement in the fields of both molecular genetics and mental retardation, there have been several papers trying to unravel the cytogenetic and molecular aspects of the 17p11.2 deletion in SMS. Smith-Magenis Syndrome is ascribed to interstitial microdeletion involving the band p11.2 of chromosome 17 and is hypothesized to be a “contiguous deletion syndrome.” This is localized to a cluster of closely linked genes that is simultaneous deleted leading to a complex phenotype that reflects contributions of multiple genes. The phenotype arises from the haploinsufficiency of multiple, functionally unrelated genes in close proximity depending on the extent of the deletion. The human genome contains “sites of instability” or “hot spots” for crossing over, breakage, and rearrangement, and these regions are shown to be vulnerable for deletions.28 The presence of such hot spots in 17p11.2 could explain the relatively high frequency of SMS as a de novo chromosomal deletion.
Studies are targeted towards the molecular definition of the “critical deletion interval” in the chromosomal band 17p11.2 and to identify “candidate genes” and “proximal/distal breakpoints” in this interval responsible for the phenotypic expression of SMS. This genetic locus at chromosome 17p near to the regions (chromosome band 17p12) is associated with the peripheral neuropathies, hereditary neuropathy with liability to pressure palsies29 and Charcot-Marie-Tooth disease Type 1A (CMT 1A).30,31 The SMS common deletion interval is estimated to contain 4–5 Mb of deoxyribonucleic acid (DNA) or approximately 100 genes based on an average gene size of approximately 50 kb.32 The SMS “critical interval” defined as “the smallest deletion necessary to give the full SMS phenotype” is approximately 1.5–2 Mb (Figure 1).
Juyal et al.33 conducted a molecular genetic study using a multifaceted approach, which combined the use of somatic cell hybrid analysis, genotyping of microsatellite markers, restriction fragment length polymorphism analysis, and fluorescence in situ hybridization to determine the molecular definition of the interval defining this microdeletion syndrome at various loci in 62 unrelated SMS patients in conjunction with 70 available unaffected parents. The results confirmed the microsatellite markers that mapped the proximal and distal deletion breakpoint. A small deletion size (<2 Mb) may go undetected as it may be at the limits of the resolution of routine cytogenetic analysis and will be detected by high-resolution karyotype and fluorescence in situ hybridization. There has been a broad range of clinical manifestations that has wide variability in both the type of clinical manifestations as well as the extent of clinical severity. The phenotypic variability and severity in SMS may correspond to the degree of deletion, as larger deletions involve more genes.
Zori et al.27 described an infant with deletion 17(p11.2p12) whose deleted chromosome was transmitted from a mosaic mother. The phenotypic findings in the mother were mild enough and would not have been tested. Therefore it is standard cytogenetic procedure that when apparent terminal deletion is identified, parental chromosomes need to be studied in order to investigate whether the abnormal chromosome is derived from a balanced parental rearrangement.
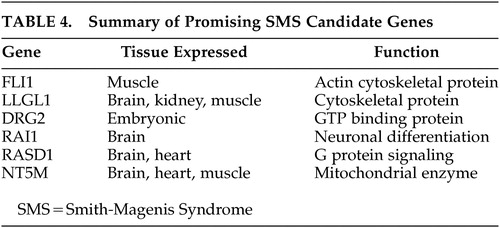
Recombination between repeated sequences at various loci of the human genome are known to give rise to DNA rearrangements associated with many genetic disorders. Low-copy repeats have been implicated as the cause of DNA rearrangement and the presence of repetitive sequences can hypothetically facilitate homologous recombination and lead to chromosomal deletions. Chromosomal deletions in humans resulting in loss of megabases of DNA in specific regions of the human genome may be explained by three mechanisms: 1) interchromosomal rearrangement, 2) intrachromosomal rearrangement, and 3) fragile site associated with an expanded trinucleotide repeat. Chen et al.34 in their work suggested that homologous recombination between flanking repeat gene cluster as a molecular mechanism that leads to the SMS microdeletion syndrome. Three copies of a low-copy-number repeat have been located within and flanking the SMS common deletion region. These chromosome-specific repetitive structures are predisposed to frequent microdeletions or microduplications that cause gross change to the genomic DNA. Potocki et al.35 reported seven unrelated patients of SMS with de novo 17p11.2 duplication that arises from unequal crossing over due to homologous recombination between flanking repeat gene clusters. Seranski et al.36 studied the genomic sequencing of the candidate deletion interval and identified one of the genes named RAI 1, which has the polymorphic CAG repeat coding for a polyglutamine stretch in the amino terminal of the protein. Bi et al.37 and Lucas et al.38 have studied the genomic organization of the 1.5-Mb SMS critical interval in order to identify possible candidate genes and described a detailed review of six promising candidate genes that map to the SMS critical region. These six genes are summarized as Table 4. Functional genomics studies many of these brain-expressed genes and their functional gene products that are of critical importance to the understanding of the complex gene-brain-behavior interrelationship.
Molecular genetic techniques in the diagnosis of SMS are becoming important tools for the prenatal diagnosis by amniocentesis and fetal anomaly ultrasound scanning.39
Conclusion
There are almost 750 known genetic disorders associated with mental retardation. Behavioral researchers must play a pivotal role in syndromic research in order to solve the behavioral half of the gene-behavior puzzle. As a part of the gene-brain-behavior axis, it is imperative to note that the advances in molecular genetics can only be interpreted in the context of a thorough and accurate behavioral examination. A multidisciplinary collaboration between behavioral researchers and geneticists is needed to appreciate the challenges in conducting behavioral phenotypic research to examine the links between genes, brain, and behavior and to derive at genotype-phenotype correlations.
Future research should continue to aim toward improving the quality of life for persons with SMS and their families. Therapeutic intervention should be directed toward drug treatment to modify aberrant behavior and improve attention span with carbamazepine therapy, speech therapy, and amelioration of sleep disturbances, all of which may contribute to cognitive and behavioral improvement. It is mandatory that every SMS patient be subjected to annual evaluation for screening multisystem abnormalities and to plan for appropriate intervention if any of these studies are abnormal. Teachers, special educators, psychologists and caregivers should be informed about all the behavioral and emotional aspects of this complex genetic syndrome, which will often make life for the child, family and teacher much easier to cope with.
ACKNOWLEDGMENTS
Dr. Shelley is a Research Fellow at the Raymond Way Neuropsychiatry Research Group, Institute of Neurology, Queen Square, London.

FIGURE 1. Full SMS Phenotype
HNPP=hereditary neuropathy with liability to pressure palsies
CMT=Charcot-Marie-Tooth disease
SMS=Smith-Magenis Syndrome
 |
 |
 |
 |
1 Smith ACM, McGarvan L, Waldstein G: Deletion of the 17 short arm in two patients with facial clefts (abstract). Am J Hum Genet 1982; 34(suppl):410Google Scholar
2 Smith ACM, McGavran L, Robinson J, et al: Interstitial deletion of (17)(p11.2p11.2) in nine patients. Am J Med Genet 1986; 24:393–414Crossref, Medline, Google Scholar
3 Greenberg F, Guzzetta V, de Oca-Luna RM, et al: Molecular analysis of the Smith-Magenis syndrome: a possible contiguous-gene syndrome associated with del(17)(p11.2). Am J Hum Genet 1991; 49:1207–1218Medline, Google Scholar
4 Greenberg F, Lewis RA, Potocki L, et al: Multi-disciplinary clinical study of Smith-Magenis syndrome (deletion 17p11.2). Am J Med Genet 1996; 62:247–254Crossref, Medline, Google Scholar
5 Willekens D, De Cock P, Fryns JP: Three young children with Smith-Magenis syndrome: their distinct, recognisable behavioural phenotype as the most important clinical symptoms. Genet Couns 2000; 11:103–110Medline, Google Scholar
6 Hodapp RM, Fidler DJ, Smith AC: Stress and coping in families of children with Smith-Magenis syndrome. J Intellect Disabil Res 1998; 42:331–340Crossref, Medline, Google Scholar
7 Smith ACM, Dykens E, Greenberg F: Sleep disturbance in Smith-Magenis syndrome (del 17p11.2). Am J Med Genet Neuropsychiatr Genet 1998; 81:186–191Crossref, Medline, Google Scholar
8 Clarke DJ, Boer H: Problem behaviours associated with deletion Prader-Willi, Smith-Magenis, and cri du chat syndromes. Am J Ment Retard 1998; 103:264–271Crossref, Medline, Google Scholar
9 Fischer H, Oswald HP, Duba HC, et al: Constitutional interstitial deletion of 17(p11.2) (Smith-Magenis syndrome): a clinically recognizable microdeletion syndrome: report of two cases and review of the literature. Klin Padiatr 1993; 205:162–166Crossref, Medline, Google Scholar
10 Colley AF, Leversha MA, Voullaire LE, et al: Five cases demonstrating the distinctive behavioural features of chromosome deletion 17(p11.2p11.2) (Smith-Magenis syndrome). J Paediatr Child Health 1990; 26:17–21Crossref, Medline, Google Scholar
11 Finucane BM, Dirrigl KH, Simon EW: Characterization of self-injurious behaviors in children and adults with Smith-Magenis syndrome. Am J Ment Retard 2001; 106:52–58Crossref, Medline, Google Scholar
12 Finucane BM, Konar D, Hass-Givler B, et al: The spasmodic upper-body squeeze: a characteristic behaviour in Smith-Magenis syndrome. Dev Med Child Neurol 1994; 36:78–83Crossref, Medline, Google Scholar
13 Vostanis P, Harrington R, Prendergast M, et al: Case reports of autism with interstitial deletion of chromosome 17(p11.2p11.2) and monosomy of chromosome 5(5pter-5p15.3). Psychiatr Genet 1994; 4:109–111Crossref, Medline, Google Scholar
14 Dykens EM, Smith ACM: Distinctiveness and correlates of maladaptive behaviour in children and adolescents with Smith-Magenis syndrome. J Intellect Disabil Res 1998; 42:481–489Crossref, Medline, Google Scholar
15 Dykens EM, Brenda M, Finucane B, et al: Cognitive and behavioural profiles in persons with Smith-Magenis syndrome. J Autism Dev Disord 1997; 27:203–211Crossref, Medline, Google Scholar
16 Udwin O, Webber C, Horn I: Abilities and attainment in Smith-Magenis syndrome. Dev Med Child Neurol 2001; 43:823–828Crossref, Medline, Google Scholar
17 Allanson JE, Greenberg F, Smith ACM: The face of Smith-Magenis syndrome: a subjective and objective study. J Med Genet 1999; 36:394–397Medline, Google Scholar
18 Quine L: Sleep problems in children with mental handicap. J Ment Defic Res 1991; 35:269–290Medline, Google Scholar
19 DeLeersnyder H, de Bois MC, Claustrat B, et al: Inversion of the circadian rhythm of melatonin in the Smith-Magenis syndrome. J Pediatr 2001; 139:111–116Crossref, Medline, Google Scholar
20 Potocki L, Glaze D, Tan DX, et al: Circadian rhythm abnormalities of melatonin in Smith-Magenis syndrome. J Med Genet 2000; 37:428–433Crossref, Medline, Google Scholar
21 DeLeersnyder H, de Bois MC, Vekemans M, et al: Beta(1)-adrenergic antagonists improve sleep and behavioural disturbances in a circadian disorder, Smith-Magenis syndrome. J Med Genet 2001; 38:586–590Crossref, Medline, Google Scholar
22 DiCicco M, Padoan R, Felisati G, et al: Otorhinolaryngologic manifestation of Smith-Magenis syndrome. Int J Pediatr Otorhinolaryngol 2001; 59:147–150Crossref, Medline, Google Scholar
23 Masuno M, Asano J, Arai M, et al: Interstitial deletion of 17p11.2 with brain abnormalities. Clin Genet 1992; 41:278–280Crossref, Medline, Google Scholar
24 Vuksanovic B, Jalal SM, Garrity JA, et al: Visual impairment due to macular disciform scars in a 20-year-old man with Smith-Magenis syndrome: another ophthalmologic complication. Am J Med Genet 1998; 80:373–376Crossref, Medline, Google Scholar
25 Chen RM, Lupski JR, Greenberg F, et al: Ophthalmic manifestations of Smith-Magenis syndrome. Ophthalmology 1996; 103:1084–1091Crossref, Medline, Google Scholar
26 Finucane BM, Jaeger ER, Kurtz MB, et al: Eye abnormalities in the Smith-Magenis contiguous gene deletion syndrome. Am J Med Genet 1993; 45:443–446Crossref, Medline, Google Scholar
27 Zori RT, Lupski JR, Heju Z, et al: Clinical, cytogenetic, and molecular evidence for an infant with Smith-Magenis syndrome born from a mother having mosaic 17p11.2p11.2 deletion. Am J Med Genet 1993; 47:504–511Crossref, Medline, Google Scholar
28 Chandley AC: On the parental origin of de novo mutation in man. J Med Genet 1991; 28:217–223Crossref, Medline, Google Scholar
29 Chance PF, Alderson MK, Leppig KA, et al: DNA deletion associated with hereditary neuropathy with liability to pressure palsies. Cell 1993; 72:143–151Crossref, Medline, Google Scholar
30 Lupski JR, Montes de Oca-Luna R, Slaugenhaupt S, et al: DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell 1991; 66:219–232Crossref, Medline, Google Scholar
31 Lupski JR: An inherited DNA rearrangement and gene dosage effect are responsible for the most common autosomal dominant peripheral neuropathy-Charcot-Marie-Tooth disease type 1A. Clin Res 1992; 40:645–652Medline, Google Scholar
32 Trask BJ, Mefford H, van den Engh G: Quantification by flow cytometry of chromosome-17 deletions in Smith-Magenis syndrome patients. Hum Genet 1996; 98:710–718Crossref, Medline, Google Scholar
33 Juyal RC, Figuera LE, Hauge X, et al: Molecular analyses of 17p11.2 deletions in 62 Smith-Magenis syndrome patients. Am J Hum Genet 1996; 58:998–1007Medline, Google Scholar
34 Chen KS, Manian P, Koeuth T, Potocki L, Zhao Q, Chinault AC, Lee CC, Lupski JR: Homologous recombination of a flanking repeat gene cluster is a mechanism for a common contiguous gene deletion syndrome. Nat Genet 1997; 17:154–164Crossref, Medline, Google Scholar
35 Potocki L, Chen KS, Park SS, et al: Molecular mechanism for duplication 17p11.2—the homologous recombination reciprocal of the Smith-Magenis microdeletion. Nat Genet 2000; 24:84–87Crossref, Medline, Google Scholar
36 Seranski P, Hoff C, Radelof U, et al: RAI1 is a novel polyglutamine encoding gene that is deleted in Smith-Magenis syndrome patients. Gene 2001; 30:69–76Crossref, Google Scholar
37 Bi W, Yan J, Stankiewicz P, et al: Genes in a refined Smith-Magenis syndrome critical deletion interval on chromosome 17p11.2 and the syntenic region of the mouse. Genome Res 2002; 12:713–728Crossref, Medline, Google Scholar
38 Lucas RE, Vlangos CN, Das P, et al: Genomic organisation of the approximately 1.5 Mb Smith-Magenis syndrome critical interval: transcription map, genomic contig, and candidate gene analysis. Eur J Hum Genet 2001; 9:892–902Crossref, Medline, Google Scholar
39 Thomas DG, Jacques SM, Flore LA, et al: Prenatal diagnosis of Smith-Magenis syndrome (del 17p11.2). Fetal Diagn Ther 2000; 15:335–337Crossref, Medline, Google Scholar

