
Does the Presentation of Creutzfeldt-Jakob Disease Vary by Age or Presumed Etiology? A Meta-Analysis of the Past 10 Years
Existing studies report a range of presenting symptoms, and there is a lack of consensus on whether etiological subtypes present differently. Some studies report that psychiatric symptoms occur in a minority of patients afflicted with sCJD, and others find differences in the prevalence of psychiatric symptoms between sCJD and vCJD. 2 – 4 On the other hand, Zerr et al. 5 suggest that there is little clinical difference between sCJD and vCJD. Other studies have delineated differences in illness presentation between young and old sCJD patients, and a recent study by Wall et al. found that 92% of their subjects had at least one psychiatric symptom during the course of their illness. 6 , 7
Variation in clinical presentation is of interest because it can influence earlier and more accurate diagnosis while potentially providing information about the pathogenesis underlying specific symptoms in CJD and other disorders. To answer some of these questions, we conducted a meta-analysis of the published literature on the presentation of the four different forms of CJD.
METHOD
We conducted a search of the literature on PubMed, restricting the search to articles published in English, those including human subjects, and those published within the last 10 years up until May 2006. Each article was then read and those that mentioned clinical signs and symptoms were retained for this study. Review and basic science articles were excluded if they did not contain clinical data. The original authors determined CJD subtype, which may or may not have been confirmed with PRNP analysis. The subcategory of CJD not otherwise specified was created for those individuals whose subtype was not disclosed by the original authors.
Once the data were obtained, related symptoms were clustered together (i.e., cerebellar dysfunction/gait disturbance). We then generated a range by using the lowest and highest values of the symptom set. Reporting the data as a range ensured that individuals would not be double-counted in the case that one person had multiple related symptoms. We used chi-square analyses to compare various CJD subtypes and age ranges. These analyses were not performed on data reported in ranges.
RESULTS
Figure 1 presents data on the number of articles reviewed. The search yielded a total of 284 articles that met inclusion criteria, of which 228 (80.2%) were either case reports or case series. Twenty-three (8.1%) articles were clinical trials, 12 (4.2%) were retrospective chart reviews, and two (0.7%) were pilot studies. All articles mentioned at least one clinical feature. Psychiatric symptoms were reported in 219 (77.1%) studies. Forty-four (15.5%) studies were concerned with nonclinical results or imaging, and 49 (17.3%) studies dealt with basic science. One hundred seventy-five studies (61.6%) were done within a single center. Sixty-two (22.8%) studies were from the United States, followed by frequency from Japan (42, 15.4%), the U.K. (41, 15%), and Germany (27, 9.9%).
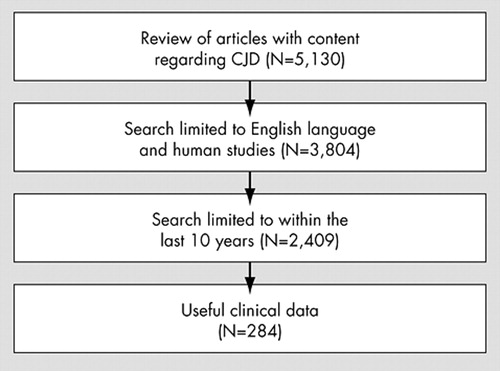
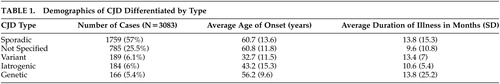
The 284 articles included in this review contained data on 3,083 people. The largest number of cases were sporadic in nature (N=1759, 57%), but in 785 cases (25.5%), CJD type was not specified. Of the subjects for whom gender was specified, there were 584 men (42.8%) and 781 women (57.2%). Many studies did not include information on gender. The average age of onset was 55.6 (SD=15.6) years and the maximum age of onset was 90 years. Table 1 shows the average age and illness duration for each CJD type. Of note, vCJD had the youngest mean age of onset (32.7 years), followed by iCJD cases (43.2 years). The average age of onset for sCJD was 60.7 years. The average length of illness also varied by CJD type with iatrogenic and unspecified cases having a shorter course than the sporadic, variant, and genetic types. Analysis of variance (ANOVA) showed a statistically significant difference in age of onset according to CJD type (p<0.0001). There was no statistically significant difference in illness duration when separated by CJD type. However, illness duration did vary by age of onset. The survival time of patients under 50 years old (average age=34.3 years) was 14.8 months (SD=18.9). In patients older than 50 (average age of 64.3 years), the survival time was shorter at 10.3 months (SD=10.1)(p=0.048).
 |
As shown in Table 2 , the most common presenting symptom among all CJD types was cerebellar/gait disturbance (22%), followed by dementia/cognitive decline/memory impairment (1.3% to 21.4%). Visual/oculomotor disturbance was present in 15% of cases, and behavior/personality change in 10.5%. The most common symptoms throughout the course of the illness were cognitive impairment, cerebellar/gait disturbance, myoclonus, and visual/oculomotor disturbance.
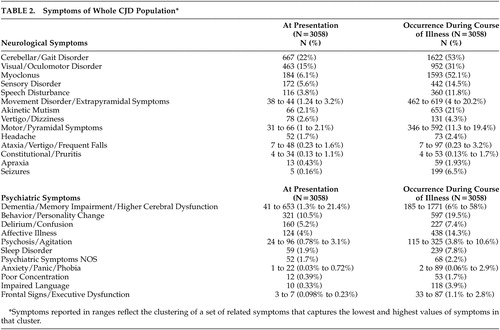 |
In a comparison between sCJD and vCJD, sCJD was found to present primarily with neurological signs, especially cerebellar/gait disturbance (16.5%, p=0.0003) and dementia/memory impairment/higher cerebral dysfunction (2.1% to 18.9%, p=0.0001), as well as behavioral/personality change (12.7%, p=0.009). In addition, sCJD was more likely to present with delirium/confusion (6.4%, p=0.0001). On the other hand, vCJD presented with more affective illness (14.8%, p=0.0001) and psychiatric symptoms not otherwise specified (11.6%, p=0.001). vCJD also presented with more sensory complaints (22.7%, p=0.0001) and motor/pyramidal symptoms (3.7%, p=0.04) compared with sCJD.
Genetic CJD presented mostly with cerebellar/gait disturbance (10.8%) and dementia/memory impairment/higher cerebral dysfunction (3.6% to 10.2%). Iatrogenic CJD presented with a higher prevalence of delirium/confusion (17.4%), possibly indicating a more compromised cognitive state at baseline. Iatrogenic CJD also commonly presented with dementia/memory impairment/higher cerebral dysfunction (13.6% to 38%) and behavior/personality changes (24.4%).
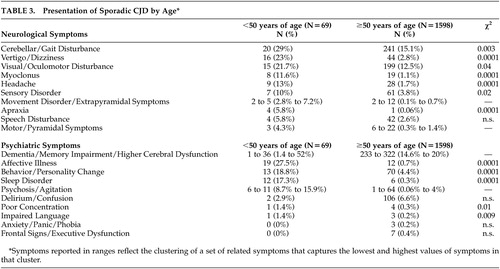
As shown in Table 3 , young (<50 years old) individuals with sCJD were more likely to present with cerebellar/gait disorders (p=0.003), visual/oculomotor disturbance (p=0.04), myoclonus (p=0.0001), sensory disorder (p=0.02), vertigo/dizziness (p=0.0001), headache (p=0.0001), and apraxia (p=0.0001), compared with older individuals (>50 years old). From a psychiatric standpoint, younger individuals were more likely to present with affective illness (p=0.0001), behavior/personality change (p=0.0001), and sleep disorder (p=0.0001). Some symptoms (e.g., dementia/memory impairment/higher cerebral dysfunction, and psychosis) could not be examined statistically because they were reported as ranges. However, young sCJD patients did have a higher percentage of dementia and psychotic symptoms upon presentation. Overall, there was a higher percentage of psychiatric presenting symptoms in the younger group of sCJD patients.
 |
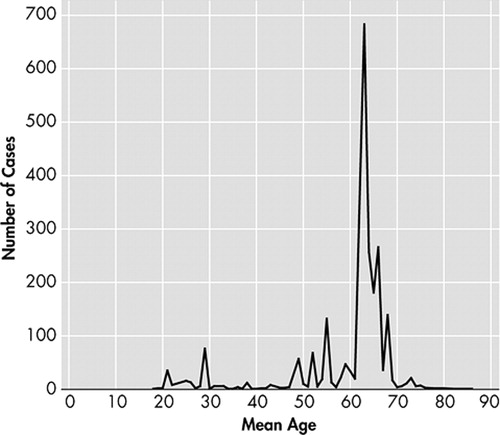
Because of these differences between young and old sCJD patients, the relationship between age and CJD types was also examined. A scatter-plot ( Figure 2 ) suggests that the age of onset is bimodally distributed with age 50 as the approximate separation point. Based on this information, we performed analyses comparing those less than 50 years of age to those older than 50 years of age. Variant CJD patients were excluded from this analysis as they have already been shown to present primarily with psychiatric symptoms. The results of the analysis can be viewed on Table 4 . Younger CJD patients were most likely to present with cerebellar/gait disturbance (50.9%, p=0.0001), visual/oculomotor disturbance (34%, p=0.0001), sensory disorder (17.8%, p=0.0001), speech disturbance (9.8%, p=0.0002), vertigo/dizziness (10.4%, p=0.0001), headache 12.9%, p=0.0001), and apraxia (2.5%, p=0.005). Though there was not a statistically significant difference between the two groups regarding behavior/personality change, young sCJD were more likely to present with affective illness (28.8%, p=0.0001), sleep disorder (20.2%, p=0.0001), and poor concentration (3%, p=0.0003). In contrast, older CJD patients were more likely to present with delirium (6.3%, p=0.04). Presenting symptoms were further differentiated based upon type and age ( Figures 3 and 4 ). Sporadic CJD patients less than 50 years of age, vCJD patients, and all young CJD types except for variant were compared to gCJD, older sCJD, and all older CJD patients except variant. Affective symptoms were more likely to present in the younger groups of patients, as were cerebellar/gait disturbance and sensory symptoms. Moreover, vCJD presented much more often with sensory symptoms when compared with the other groups (22.7%, p=0.0001).
 |
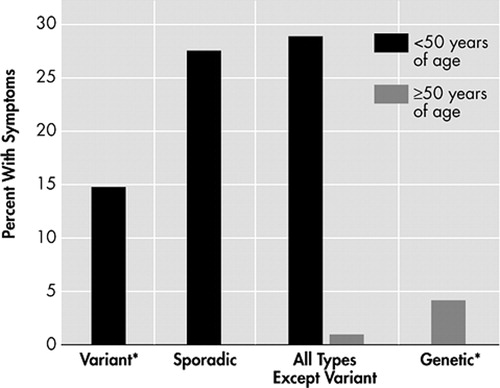
* There were 2 variant cases >50 years and 16 genetic cases <50 years.
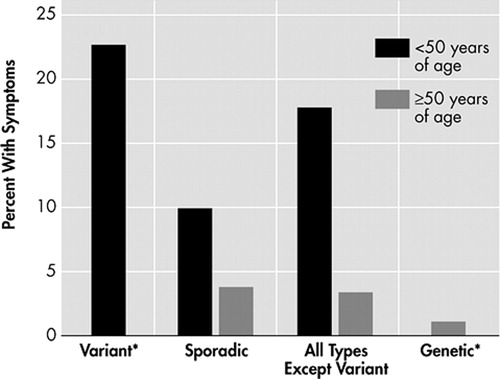
* There were 2 variant cases >50 years and 16 genetic cases <50 years.
Psychosis/agitation was found to be more prominent in young CJD patients (8% to 8.5%). Movement disorder was likewise more common in young CJD patient presentations (3% to 12.9%). Also of note, dementia/memory impairment/higher cerebral dysfunction tended to present more in all young CJD types (16% to 28.8%). In fact 1.4% to 52% of young sCJD patients presented with dementia/memory impairment/higher cerebral dysfunction. Other psychiatric symptoms, such as sleep disorder and anxiety/panic/phobia, were also more prominent within the younger group of patients (0.6% to 2.4%).
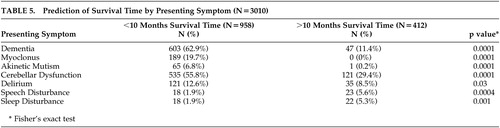
Certain presenting symptoms were predictive of survival time ( Table 5 ). For example, those individuals with a survival time of less than 10 months were more likely to present with dementia, myoclonus, akinetic mutism, cerebellar dysfunction, and delirium. This group’s average age was 61.2 years (SD=8.1). Interestingly, patients who survived longer than 10 months reported presenting symptoms of speech and sleep disorders. The average age of this group was 51 years (SD=12.4)
 |
DISCUSSION
This meta-analysis confirms the view that the presentation of CJD varies by presumed etiology. Variant CJD and iCJD patients have an earlier age of onset. CJD not otherwise specified and iCJD had the shortest survival times from disease onset. Sporadic CJD was more likely to present with “neurological” symptoms while vCJD commonly presented with affective illnesses and psychiatric symptoms not otherwise specified. Sensory complaints were also more common in vCJD patients. Genetic CJD presented mostly with cerebellar/gait disturbance and dementia/memory impairment/higher cerebral dysfunction, whereas iCJD presented with a higher prevalence of delirium/confusion which could represent premorbid cognitive compromise from prior neurological injuries requiring dural grafts. All CJD types presented mostly with cerebellar or gait disturbance, followed by dementia/cognitive decline/memory impairment. Cerebellar/gait disturbance, myoclonus, and visual/oculomotor disturbance were the most common symptoms throughout the course of illness.
This meta-analysis also supports the contention that age rather than etiological subtype explains some differences in symptom presentation. Younger age was associated with a higher likelihood of presenting with cerebellar/gait disturbance, visual/oculomotor disturbance, sensory disorder, speech disturbance, seizures, vertigo/dizziness, headache, and apraxia, regardless of etiology. Additionally, younger patients had higher presentation rates of affective illness, sleep disorder, and poor concentration. Older patients were more likely to present with delirium, perhaps due to other non-CJD related brain disorders or lower cognitive reserves. The similarities in presenting symptoms of sCJD and gCJD patients may also be a consequence of age as these subtypes tend to be older. It is also possible that some of the reported sCJD cases were actually gCJD, which may have erroneously lowered the average age of sCJD cases. Nevertheless, we found that neuropsychiatric symptoms are common in specific age groupings across etiological subtypes.
The longer duration of illness in patients less than 50 years of age could reflect less premorbid neurological and medical disease. This, in turn, could lead to a protracted course of illness. Although there appears to be a difference between the etiological causes of illness, symptom presentation could, in fact, be attributable to age. For example, sCJD patients tend to be older than vCJD patients, so it is possible that age may be a better predictor of symptom presentation instead of etiological type. Also, there was a striking difference in the rate of affective illness between the two age groups in that younger patients were much more likely to present with a mood disorder. This difference was preserved across CJD types and may very well be a result of the age of presentation as opposed to type. There are several plausible explanations for the findings that younger CJD patients have higher rates of certain presenting symptoms. The increased rate of psychiatric illness, namely affective illness, may reflect an encephalopathic process that precedes coarse brain disease. Hence, younger patients with higher cognitive reserves would present with psychiatric symptoms prior to neurological ones. However, young age of onset also predicted certain neurological symptoms. Because younger patients have a higher cognitive reserve, this may also necessitate serious neurological injury prior to presentation, leading to the higher percentages of neurological symptoms in younger patients. Another possibility is reporting bias within the sample. Young patients with CJD are rare, and this may have led to an overreporting of symptoms and cases as compared to later onset CJD cases. This would result in overreporting of both neurological and psychiatric symptoms for the younger CJD population.
The examination of presenting symptoms with regard to survival time is also interesting. As previously mentioned, however, one must consider the age range of the two survival groups depicted in Table 5 . Individuals with a younger age of onset generally live longer than those above age 50. Those individuals with the longest survival times were also younger and, in fact, presented similarly to the young age of onset group. The longer survival time cases generally presented with speech and sleep disorders. Another plausible explanation is that those individuals with shorter survival times were more likely to have been reported further into their disease course. This would result in more dementia, akinetic mutism, and myoclonus, which are symptoms commonly seen in more advanced CJD.
There are several limitations to this study. As in any meta-analysis, the quality of the study is dependent on the data gleaned from the literature. Because neurologists generally report CJD cases, it is possible that psychiatric symptoms were underreported. However, the majority (77%) of studies included psychiatric symptoms. Additionally, the young average age of onset for sCJD may be a result of clinicians reporting atypically young sCJD patients, which would bias the data. Because we carried over the subtype designated by the original authors, it is possible that some sCJD cases were not confirmed with PRNP analysis. Without molecular confirmation, it is possible that some reported sCJD cases were actually gCJD cases, which have a younger age of onset. Such a phenomenon would falsely lower the age of onset in sCJD. Another drawback to this study is that genotypes were not explored. This is relevant because different molecular subtypes of sCJD present differently and have varying age of onset, survival times, and even neuropathology. 8 For example, Collins et al. 9 reported an average age of onset of 47.2 years for the VV1 subtype. Thus, the data may reflect clinical characteristics of certain subtypes while appearing to be an age-related phenomenon. One argument that contradicts this occurrence is that there was an age-related effect throughout all CJD subtypes, not just sCJD. Also, it is possible that atypical sCJD variants are overreported in the literature, which would bias our sCJD data.
CONCLUSIONS
In conclusion, presenting symptoms differ both by CJD type and by age of onset. CJD type predicts certain symptoms, such as sensory disorder. Age, on the other hand, is also a vital factor in symptom presentation. Symptoms such as affective illness appear to be largely influenced by age rather than by subtype. Further research could examine the various forms of CJD and presenting symptoms in a prospective manner. Another fruitful endeavor would be the continued investigation of symptom presentation in relation to molecular subtype. Finally, studying the progression of CJD symptoms may offer insight into other neurological and psychiatric illnesses.
1 . Gambetti P, Parchi P, Chen S: Hereditary Creutzfeldt-Jakob disease and fatal familial insomnia. Clin Lab Med 2003; 23:43–64Google Scholar
2 . Zeidler, M, Johnstone EC, Bamber RW, et al: New variant Creutzfeldt-Jakob disease: psychiatric features. Lancet 1997; 350:908–910Google Scholar
3 . Will RG, Matthews WB: A retrospective study of Creutzfeldt-Jakob disease in England and Wales 1970–79. J Neurol Neurosurg Psychiatry 1984; 47:134–140Google Scholar
4 . Brown P, Cathala F, Sadowsky D, et al: Creutzfeldt-Jakob disease in France: II, clinical characteristics of 124 consecutive verified cases during the decade 1968–1977. Ann Neurol 1979; 6:430–437Google Scholar
5 . Zerr I, Weidehaas K, Lantsch A: Psychiatric symptoms in sporadic Creutzfeldt Jakob Disease. J Neuro Sci 1997; 150(Suppl 1):S191-S192Google Scholar
6 . Boesenberg C, Schulz-Schaeffer WJ, Meissner B, et al: Clinical course in young patients with sporadic Creutzfeldt-Jakob disease. Ann Neurol 2005; 58:533–543Google Scholar
7 . Wall CA, Rummans TA, Aksamit AJ, et al: Psychiatric manifestations of Creutzfeldt-Jakob disease: a 25-year analysis. J Neuropsychiatry Clin Neurosci 2005; 17:489–495Google Scholar
8 . Hill AF, Joiner S, Wadsworth JD, et al: Molecular classification of sporadic Creutzfeldt-Jakob disease. Brain 2003; 126(Pt 6):1333–1346Google Scholar
9 . Collins SJ, Sanchez-Juan P, Masters CL, et al: Determinants of diagnostic investigation sensitivities across the clinical spectrum of sporadic Creutzfeldt-Jakob disease. Brain 2006; 129(Pt 9):2278–2287Google Scholar

