
GAD65 Autoantibodies in Kleine-Levin Syndrome
To the Editor: Kleine-Levin syndrome (KLS), or Sleeping Beauty syndrome, is a rare disorder that mainly affects adolescents and is characterized by recurring episodes of severe hypersomnia, cognitive impairment, apathy, derealization, and psychiatric and behavioral disturbances.1
Slightly over 50% of KLS patients have hyperphagia, are hypersexual (mainly males), or have depressed mood (mainly females), and 30% become anxious, delusional, and have hallucinations.1 Overall, males are more frequently affected than females.1 At the onset of an episode, the patient becomes drowsy and sleeps for most of the day and night, waking only to eat or use the bathroom. Most are bedridden, tired, and uncommunicative even when awake.2
The exact cause of KLS is not known, however, the first episodes are generally triggered by infection.3 Recent studies suggest genetic and autoimmune etiologies.4 Serum white blood cell counts and markers of inflammation are within normal limits during and between episodes.1 However, postmortem studies have shown perivascular infiltrates in the amygdala and temporal lobe and signs of encephalitis in the thalamus and hypothalamus of KLS patients.1
Autoantibodies against brain proteins are associated with neurobehavioral problems.5 These autoantibodies may cross the blood brain barrier during inflammation and exposure to environmental agents.6 Antibodies against molecules regulating brain function may disturb the excitatory and inhibitory balance in KLS.7 However, no studies thus far have reported on the levels of autoantibodies against any brain proteins in patients diagnosed with KLS. In the present study, we have examined autoantibodies against glutamic acid decarboxylase isoform 65 (GAD65), an enzyme that synthesizes inhibitory neurotransmitter γ-aminobutyric acid, in the serum of a KLS patient.
Case Report
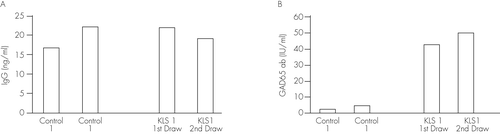
The patient is an 11-year-old African American boy, with three recent admissions with similar presentations to the University Child Psychiatry unit during a 3-month period. During those admissions, patient presented with abnormal hand movements, perseverative speech, hypersexuality, hypersomnia, social withdrawal, and decreased appetite. Patient reported visual hallucinations during this time period as well. Patient also became defiant at school and had blank staring episodes in class. The recurring pattern of episodes and symptoms met criteria for KLS. Patient’s EEG was normal, and CT without contrast was normal, antistreptolysin O (ASO) titer was barely positive with 200 (0–199) IU/ml and pediatric acute-onset neuropsychiatric syndrome (PANDAS) was ruled out. Antibodies against N-methyl-D-aspartate (NMDA) receptors were estimated commercially (ARUP Laboratories, Salt Lake City, UT) by indirect immunofluorescence assay. Antibodies against NMDA receptors (NMDAR) in the blood samples of the patient were negative, ruling out anti-NMDAR encephalitis. The patients did not suffer from diabetes and had normal blood glucose levels. Control subjects were 2 healthy males of the same age without neuropsychiatric or medical diagnoses (including diabetes). Venous blood was collected from the arm of each subject after consent was obtained. The sample was collected from the patient diagnosed with KLS twice at an interval of 2 months. Venous blood from two sex- and age-matched normal subjects was collected only once. Blood collection and enzyme-linked immunosorbent assays (ELISA) for total immunoglobulin G (IgG) content and antibodies against GAD65 were performed as reported previously.5
Total IgG content was not different among subjects (Figure 1A). The antibodies against GAD65 were approximately nine-fold higher in the serum of the patient diagnosed with KLS compared with controls (Figure 1B).
Discussion
The findings support that patients with KLS may have immunological abnormalities affecting the GABAergic system as previously suggested.8 The absence of any difference in the total IgG content and the large difference in the antibodies against GAD65 between the patient and controls support altered levels of specific Immunoglobulin (Ig) isoforms in patients with KLS. Additional studies are required to ascertain this possibility.
Antibodies cross the blood brain barrier and penetrate cells.9,10 Therefore, autoantibodies against GAD65 may penetrate neurons and disturb the GABAergic system,11 especially in the hypothalamic nuclei that are enriched with GABAergic neurons12 leading to core manifestations of KLS such as hyperphagia,13 hypersomnia,14 and hypersexuality.15 Future studies examining the cross-reactivity of serum and specifically of GAD65 autoantibodies from KLS patients with the rodent brain sections may provide information on the regional and cellular targets of these autoantibodies in the brain.
The findings presented here require replication in other KLS patients and further studies in rodent models to understand any immunological role in the neuropathology and behavioral abnormalities in KLS. Moreover, the fact that GAD65 auto antibody is found in several other neurobehavioral problems, examining arrays of antibodies including the GAD65 antibody in the blood as well as in the cerebrospinal fluid of KLS patients may be required to determine the repertoire of autoantibodies and their origin, and to ascertain the existence of biomarkers of this disease.
1 : Diagnosis, disease course, and management of patients with Kleine-Levin syndrome. Lancet Neurol 2012; 11:918–928Crossref, Medline, Google Scholar
2 : [Kleine-Levin syndrome: state of the art]. Rev Neurol (Paris) 2008; 164:658–668Crossref, Medline, Google Scholar
3 : First attack of Kleine-Levin syndrome triggered by influenza B mimicking influenza-associated encephalopathy. Intern Med 2012; 51:1605–1608Crossref, Medline, Google Scholar
4 : Kleine-Levin syndrome: an autoimmune hypothesis based on clinical and genetic analyses. Neurology 2002; 59:1739–1745Crossref, Medline, Google Scholar
5 : Presence of GAD65 autoantibodies in the serum of children with autism or ADHD. Eur Child Adolesc Psychiatry 2012; 21:141–147Crossref, Medline, Google Scholar
6 : Uptake of systemically administered human anticerebellar antibody by rat Purkinje cells following blood-brain barrier disruption. Acta Neuropathol 1995; 89:341–345Crossref, Medline, Google Scholar
7 : Dual impairment of GABAA- and GABAB-receptor-mediated synaptic responses by autoantibodies to glutamic acid decarboxylase. J Neurol Sci 2003; 208:51–56Crossref, Medline, Google Scholar
8 : Monozygotic twins concordant for Kleine-Levin syndrome. BMC Neurol 2012; 12:31Crossref, Medline, Google Scholar
9 : Anti-amyloid beta protein antibody passage across the blood-brain barrier in the SAMP8 mouse model of Alzheimer’s disease: an age-related selective uptake with reversal of learning impairment. Exp Neurol 2007; 206:248–256Crossref, Medline, Google Scholar
10 : Broken dogma: penetration of autoantibodies into living cells. Immunol Today 1996; 17:163–164Crossref, Medline, Google Scholar
11 : A pathogenetic model of autism involving Purkinje cell loss through anti-GAD antibodies. Med Hypotheses 2008; 71:218–221Crossref, Medline, Google Scholar
12 : Glutamatergic and GABAergic innervation of human gonadotropin-releasing hormone-I neurons. Endocrinology 2012; 153:2766–2776Crossref, Medline, Google Scholar
13 : An immunological basis of hyperphagia driven by GABAergic dysfunction in Prader-Willi Syndrome. Med Hypotheses 2012; 78:462–464Crossref, Medline, Google Scholar
14 : Localization of the brainstem GABAergic neurons controlling paradoxical (REM) sleep. PLoS One 2009; 4:e4272Crossref, Medline, Google Scholar
15 : Pharmacological treatment for Kleine-Levin Syndrome. Cochrane Database Syst Rev 2009; 2:CD006685Crossref, Medline, Google Scholar

