
Regional Alpha-Synuclein Aggregation, Dopaminergic Dysregulation, and the Development of Drug-Related Visual Hallucinations in Parkinson’s Disease
 |
Although the clinical and epidemiological features of visual hallucinations in Parkinson’s disease have been extensively reviewed, 2 their etiopathogenesis remains open to debate. There is a critical gap in the knowledge base that centers on neurodegenerative alterations and neurotransmitter system deficiencies in Parkinson’s disease patients with visual hallucinations and other psychotic symptoms. This seriously limits therapeutic efficacy and may prevent the successful development of novel interventions like neurogenic stem cells grafting 8 , 9 and gene therapy using viral vector-mediated delivery of glial cell-derived neurotrophic factor in the substantia nigra and the striatum. 10 – 12
In this review the current knowledge on the potential impact of the regional neurodegenerative process of Parkinson’s disease in the development of visual hallucinations as well as the possible role of the dopamine transporter and dopamine receptors is summarized.
The Role of Alpha-Synuclein
Alpha-synuclein is a marker of neurodegeneration in Parkinson’s disease. 13 It is a major constituent of Lewy bodies and Lewy neurites, 14 – 16 which have been found in higher densities in temporolimbic areas in patients with visual hallucinations. 17 Increased alpha-synuclein burden in temporolimbic and mesocorticolimbic areas may therefore be one of the key factors related to the development of visual hallucinations and other psychotic symptoms in Parkinson’s disease patients.
The synucleins are a family of soluble presynaptic proteins that are abundant in neurons and include alpha-synuclein, beta-synuclein and gamma-synuclein. 18 , 19 The function of alpha-synuclein, not yet fully clear, appears to involve maintenance and transport of synaptic vesicles 20 , 21 and participation in neuronal function through multiple interactions with other proteins and through its involvement in lipid transport and membrane biogenesis. 22 Mutations in the alpha-synuclein gene leading to abnormal protein folding 23 , 24 or protein overexpression 25 have been shown to cause familial Parkinson’s disease. Pathological inclusions of alpha-synuclein have been reproduced in several animal models. 26 – 28 One example is in the drosophila melanogaster, where alpha-synuclein overexpression results in degeneration of dopaminergic neurons and motor deficits. 27 Therefore, several lines of evidence strongly support that altered alpha-synuclein function is related or can trigger the degeneration of dopaminergic neurons.
Another function of the protein alpha-synuclein is the modulation of dopamine transporter, thus maintaining the synaptic buffering of dopamine. Dopamine transporter acts to terminate dopaminergic neurotransmission by reaccumulation of dopamine into presynaptic neurons. It thus plays a central role in the spatial and temporal buffering of released dopamine and key roles in its recycling. 29 Disruption of this function of alpha-synuclein may result in abnormal intracellular and extracellular dopamine content, which may represent an alternative route leading to degeneration of the presynaptic dopaminergic nerve terminals. 13
The substantia nigra is among the predilection sites of the Parkinson’s disease-related Lewy bodies and Lewy neurites. These inclusion bodies, together with the subsequent loss of its neuromelanin-containing dopaminergic projection neurons, constitute the main pathological features of the disorder. 30 Although the substantia nigra is an important focal point of the pathology, the underlying process is by no means confined to this midbrain nuclear complex. A variety of extranigral regions, especially those assigned to the limbic system (including the amygdala), are among the hardest hit targets of the Parkinson’s disease-related lesions. 31 – 33 In a clinicopathologic study 17 comparing patients with dementia with Lewy bodies and Parkinson’s disease with and without dementia, a striking association between the distribution of temporal lobe Lewy bodies and well-formed visual hallucinations was reported. Patients with well-formed visual hallucinations had high densities of Lewy bodies in the amygdala and parahippocampus, with early hallucinations relating to higher densities in parahippocampal and inferior temporal cortices. The results of this study relate the distribution of temporal lobe Lewy bodies (and therefore indirectly the alpha-synuclein burden) to the presence of visual hallucinations. Higher average Lewy bodies densities have been previously observed in paralimbic cortices in patients with visual hallucinations compared with those without. 34
The Role of the Ventral Tegmental Area, the Nucleus Accumbens and the Amygdala
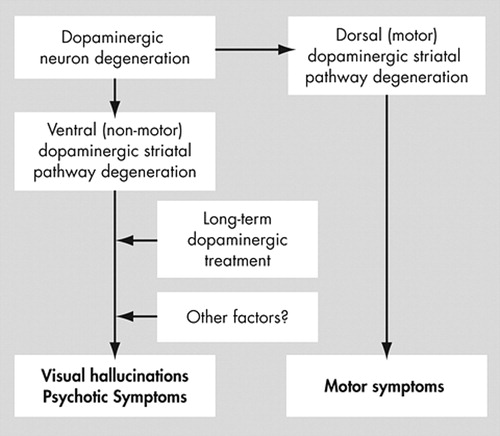
The pathological hallmark of Parkinson’s disease is the degeneration of dopamine neurons of the ventral mesencephalon. This neurodegenerative process is selective, affecting specific neuronal subpopulations, whereas other neuronal groups are relatively spared. 35 , 36 In general, the mesotelencephalic dopaminergic system may be divided into two major parts: the dorsal, nigrostriatal, part, which is mainly responsible for motor programming; and the ventral part, which extends from the ventral tegmental area through the ventral striatum either to the limbic (mesolimbic system) or frontal cortex (mesocortical system), and which is implicated in thought and behavioral programming. While disruption of the dorsal (nigrostriatal) dopamine pathway leads to the motor symptoms commonly seen in Parkinson’s disease, disruption of the ventral mesocorticolimbic dopamine system may be the key factor in the development of psychotic symptoms (mainly visual hallucinations). A schematic representation of this division is provided in Figure 1 .
Ventral Tegmental Area
The ventral tegmental area is a region in the mesencephalon which is dorsomedial to the substantia nigra and ventral to the red nucleus. The mesocortical and mesolimbic dopaminergic systems originate here, including important projections to the ventral striatum and the nucleus accumbens. Dysfunction of the neurons with their cell bodies in the ventral tegmental area and projections to the prefrontal cortex and limbic system have been implicated in psychotic states. 37 In Parkinson’s disease, ventral tegmental area neurons are depleted from 36% to 55% of control values. 38
The midbrain dopamine system can be divided into two groups of cells based on chemical characteristics and connectivity. The dorsal tier neurons, which include the dorsal substantia nigra pars compacta and the ventral tegmental area, are calbindin-positive, and project to the nucleus accumbens. The ventral tier neurons are calbindin-negative and project to the sensorimotor striatum. 39 In general, neurons of the ventral tier of the substantia nigra pars compacta are more vulnerable in Parkinson’s disease patients, followed by those of the dorsal tier, with relative sparing of the medial substantia nigra and pars lateralis. 40 In Parkinson’s disease patients with visual hallucinations and other psychotic symptoms, dorsal tier areas and especially the ventral tegmental area may be more affected when compared to patients without. The ventral tegmental area may act in the development of psychotic symptoms in a way similar to the substantia nigra in the development of motor symptoms.
Nucleus Accumbens
The concept of the ventral striatum was originally developed in the classic paper by Heimer and Wilson 41 that describes the relationship between the nucleus accumbens and the olfactory tubercle in rats. The authors showed that striatal-like and pallidal-like elements of the olfactory tubercle constitute a ventral continuation of the striatum, and, therefore, together with the nucleus accumbens, these structures should be referred to as the ventral striatum. Since then, the ventral striatum and its connections have been at the center of the reward circuit, and, as such, have been a research focus of the neurobiological mechanisms underlying drug addiction, and mental and thought disorders. 42
The nucleus accumbens is that part of the ventral striatum that has long been associated with the limbic system or with that group of structures thought to mediate motivational and emotional responses to environmental stimuli. This association is based on, among other things, its afferent connections from the amygdala and prefrontal cortical areas (both involved in mechanisms of reward and positive reinforcement). 39 The nucleus accumbens and its related circuits are also implicated in thought, attention and cognition. Dysfunction in the information processing in the nucleus accumbens may lead to the development of visual hallucinations. 43 , 44 Nucleus accumbens receives afferent input from brain regions presumably involved in the pathogenesis of schizophrenia, including the ventral tegmental area, prefrontal cortex, the hippocampus and the amygdala. 45 It sends projections to the ventral pallidum, which in turn sends a major projection to the mediodorsal nucleus of the thalamus, which is interconnected with the prefrontal cortex and regulates its activity. 46 , 47 There is evidence that both hippocampal and amygdalar input to the nucleus accumbens can affect its capability of processing and transferring information from prefrontal afferents to the ventral pallidum and the thalamocortical system. 48 In the normal functioning brain, the balance of inputs from cortical and mediotemporal structures to the nucleus accumbens is used to prepare, initiate and prevent selected thoughts and behaviors. 49 As such, the nucleus accumbens acts as a subcortical integrative hub, connecting forebrain and limbic structures and controlling the flow and processing of information. 50 Structural (neurodegenerative) damage and neurochemical imbalance in this sensitive striatal area may precipitate the genesis of visual hallucinations and possibly other psychotic phenomena seen in Parkinson’s disease. In summary, the involvement of the nucleus accumbens in the generation of visual hallucinations and other psychotic symptoms may be as important as the function of the internal part of the globus pallidus in the generation of motor symptoms.
Amygdala
The amygdala has long been suspected to contribute to psychotic symptoms because its stimulation (often by temporal lobe epilepsy) could provoke positive symptoms like hallucinations. 51 , 52 A clinicopathological analysis of demented patients with dementia with Lewy bodies revealed high amygdala Lewy bodies densities in patients who had well-formed visual hallucinations during life. 17 In another study of nondemented Parkinson’s disease patients, 53 a similar correlation was observed, with the subregion involved localized to the basolateral nucleus. Selective basolateral neuronal loss has been also reported in temporal lobe epilepsy, 54 where memory-like hallucinations and feelings of deja-vu were evoked from amygdala stimulation in 73% of cases. 55
Recent experiments have implicated the amygdala in visual dysfunction by showing its important role in the integration of parallel visual systems. 56 , 57 The amygdala appears to be the final processing center for the ventral stream of the cortical visual system subserving conscious visual identification and discrimination 58 and the extrageniculostriate (colliculo-thalamo-amygdala) visual system, which is related with automatic, nonconscious emotional visual processing (blindsight). 58 Dopamine receptor activation in the basolateral nucleus removes prefrontal cortex-induced suppression of neuronal output and enhances activity driven by sensory inputs. 59 The increased alpha-synuclein burden in the basolateral amygdaloid nucleus is likely to disrupt the ability of the amygdala to integrate coordinated behavioral responses between the two visual systems. This, in association with dopamine replacement therapies, may precipitate the visual hallucinations experienced by the patients with Parkinson’s disease.
Dopaminergic System Involvement in the Development of Visual Hallucinations in Parkinson’s Disease
Catecholamines of the midbrain, especially dopamine, are known to be involved in the pathophysiology of psychiatric disease and especially in schizophrenia. 60 Furthermore, classical neuroleptic drugs used in the treatment of schizophrenia act primarily on receptors for dopamine. 61 Visual hallucinations not only are drug-related in the majority of cases, but lowering or withdrawal of dopamine stimulation usually leads to improvement of symptoms. If the above adjustments fail to eliminate or sufficiently alleviate visual hallucinations, a trial of antipsychotics should be considered. 62 , 63 Attention must be paid to avoid exacerbation of parkinsonism, since all of the antipsychotics have the potential capacity to block striatal dopamine D2 receptors. The best choices at present are atypical antipsychotics, clozapine and quetiapine, which can reduce visual hallucinations and other psychotic symptoms without worsening parkinsonism. 64
Dopamine Transporter
Dopamine transporter is an important molecule in the pathogenesis of motor and nonmotor symptoms in Parkinson’s disease. 65 Human dopamine transporter is a 620 amino acid, single-subunit membrane protein with 12 membrane spanning regions and a large second extracellular loop. 66 It is most selectively expressed by the dopamine neurons most damaged in Parkinson’s disease. The activity and levels of dopamine transporter expression provide vital determinants of dopamine function. Dopamine transporter acts to terminate dopamine neurotransmission by reaccumulation of dopamine into presynaptic neurons. It thus plays a central role in its recycling and its temporospatial buffering. 29 Reuptake activity has a profound effect on dopaminergic neurotransmission and its biochemical, physiological, and behavioral functions.
There are regional variations in the expression of dopamine transporter. Dopamine transporter is relatively low throughout the ventral striatum. This pattern is consistent with the fact that the dorsal tier dopamine neurons express relatively low levels of mRNA for the dopamine transporter compared to the “ventral tier.” 67 Animal data indicate that dopamine transporter levels are also related to chronic exposure to dopaminergic drugs that can alter, by an as yet unknown mechanism, striatal levels of dopamine transporter without any change in the number of dopamine nerve terminals. However, in such studies the direction of the change is not always consistent. 68 Striatal dopamine transporter levels following L -dopa treatment have been reported to be unchanged, 69 – 75 increased, 72 , 76 and reduced 77 in both animal and human studies.
Alterations in dopamine transporter expression may prove important for understanding the neurochemical basis for visual hallucinations and other psychotic symptoms in Parkinson’s disease. Support for the involvement of dopamine transporter is also provided by high affinity binding of psychotherapeutic drugs (mazindol, nomifensine, and tricyclic antidepressants), drugs of abuse (including cocaine, d -amphetamine, and phencyclidine), and neurotoxins such as 6-hydroxydopamine and 1-methyl-4-phenylpyridinium (the active metabolite of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine [MPTP]) to dopamine transporter. 78
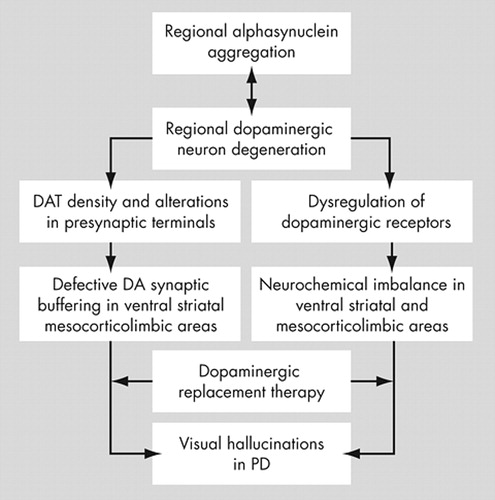
The reduction in striatal dopamine transporter probably reflects dopamine neuronal loss (presynaptic terminal loss) that contributes to some of the negative clinical outcomes in patients with Parkinson’s disease. Any loss of the ability to prevent marked changes in synaptic dopamine concentration following replacement treatment with L -dopa could increase the likelihood of developing long-term, drug-induced response fluctuations. 79 We believe that these fluctuations may be either motor (on-off phenomena and dyskinesias) or nonmotor (positive psychotic symptoms like visual hallucinations—thought fluctuations). During the initial stages of the disease, before the severe dopaminergic nerve terminal damage, the coordinated regulation of dopamine transporter and alpha-synuclein maintains some synaptic buffering of dopamine. 13 With disease progression the modulatory action of alpha-synuclein is partially lost and there is further reduction in presynaptic dopaminergic terminals, allowing the genesis of mild wearing-off symptoms. With severe neuronal and dopamine transporter loss the synaptic buffering ability is also lost contributing to visual hallucinations and other psychotic symptoms development. However, one should avoid simplistic explanations since the pathogenesis of these symptoms is far more complex and multifactorial (a review of the proposed involvement of the dopaminergic system is provided in Figure 2 ).
Dopamine Receptors
dopamine plays an important, and often necessary, role in a wide variety of behaviors and functions, ranging from movement to thought and emotion, sensitization to addiction, and development of plasticity. In part, this diversity reflects the organization of the multiple types of dopamine receptors. As all antipsychotics and antiparkinsonian agents act predominantly through the D2 subfamily of receptors, it is important to understand what functions the subtypes might mediate. This is particularly true since these receptor subtypes are differentially expressed in regions of the human brain, providing a rationale for how they could mediate different actions of dopamine. 80
An understanding of how dopaminergic receptors act in the striatum requires familiarity with its basic organization. As initially postulated, 41 it has now been convincingly demonstrated that there are parallel efferent circuits originating from the dorsal and ventral striatum with analogous “direct” and “indirect” pathways. 81 “Direct” pathway neurons project to the internal segment of the globus pallidus and substantia nigra pars reticulata, and express substance P and preprotachykinin mRNA. “Indirect” pathway neurons project to the external segment of the globus pallidus and subthalamic nucleus and express enkephalin (preproenkephalin mRNA). “Direct” (striatonigral) and “indirect” (striatopallidal) output neurons preferentially express D1 and D2 receptors, respectively. The influences of the direct and indirect pathways are considered to be opposing, and dopamine tonically modulates these two pathways via inhibition of the indirect pathway through the D2 receptor and via excitation of the direct pathway through the D1 receptor. Similar “direct” and “indirect” pathways that might be involved in visual hallucinations and other psychotic symptoms in Parkinson’s disease have been identified in the limbic striatum. Neurons within the nucleus accumbens project primarily to the ventral pallidum, with the core of the nucleus accumbens projecting to the lateral ventral pallidum, which sends projections to the subthalamic nucleus and substantia nigra pars reticulata. The shell of the nucleus accumbens projects to the medial ventral pallidum, which provides efferents to the mediodorsal thalamus. 82 As cells in the shell of the nucleus accumbens coexpress D 1 mRNA and Sub P and the cells in the core of the nucleus accumbens preferentially express D2 mRNA and ENK, 83 they are presumed to modulate direct and indirect ventral striato-pallidal-thalamo pathways, respectively. These “direct” and “indirect” limbic striatal pathways may be related to the development of visual hallucinations and other psychotic symptoms in Parkinson’s disease in a way similar to the involvement of the motor striatal “direct” and “indirect” pathways.
There are several studies (in both Parkinson’s disease animal models and patients) on the involvement of the striatal dopaminergic receptors (dorsal and ventral parts) in Parkinson’s disease. A recent report in nonhuman primates (MPTP treated monkeys) demonstrated up-regulation of both D1 and D2 dopamine receptors in the denervated striatum. 84 Consistent with these measurements of dopamine receptor protein levels, others have reported increased dopamine receptor binding in MPTP-treated monkeys and cats, as well as in Parkinson’s disease patients. 85 – 90 Striatal D2 binding has been shown to be increased in unilaterally lesioned monkeys on the denervated as compared to the control side. 91 , 92 All of these studies demonstrate that nigrostriatal dopamine denervation caused either by the neurodegenerative process of Parkinson’s disease or by selective lesioning causes a significant compensatory up-regulation of striatal dopamine receptors (denervation supersensitivity). When supersensitive dopamine receptors are stimulated by dopaminergic supplementation therapy (standard treatment in Parkinson’s disease), the delicate neurochemical balance of the nucleus accumbens may be disrupted leading to psychotic symptom development ( Figure 2 ).
Summary
The dopaminergic system seems to have a key role in the development of visual hallucinations in Parkinson’s disease via several mechanisms; however, their exact etiology remains unknown. The major event proposed in this review is the dysregulation of the ventral dopaminergic pathway that occurs with disease progression and is accompanied by regional increase of alpha-synuclein burden. Replacement therapy ( L -dopa) stimulating the supersensitive dopamine receptors of the dysregulated ventral pathway may allow the genesis of visual hallucinations. The schematic overview for the proposed role of the dopaminergic system in the development of visual hallucinations in Parkinson’s disease is shown in Figure 2 .
1. Tanner CM, Vogel C, Goetz CG: Hallucinations in Parkinson’s disease: a population study. Ann Neurol 1983; 14:136Google Scholar
2. Papapetropoulos S, Mash DC: Psychotic symptoms in Parkinson’s disease: from description to etiology. J Neurol 2005; 252:753–764Google Scholar
3. Graham JM, Grunewald RA, Sagar HJ: Hallucinosis in idiopathic Parkinson’s disease. J Neurol Neurosurg Psychiatry 1997; 63:434–440Google Scholar
4. The Parkinson Study Group: Low-dose clozapine for the treatment of drug-induced psychosis in Parkinson’s disease. N Engl J Med 1999; 340:757–763Google Scholar
5. Graham JM, Sussman JD, Ford KS, et al: Olanzapine in the treatment of hallucinosis in idiopathic Parkinson’s disease: a cautionary note. J Neurol Neurosurg Psychiatry 1998; 65:774–777Google Scholar
6. Goetz CG, Stebbins GT: Risk factors for nursing home placement in advanced Parkinson’s disease. Neurol 1993; 43:2227–2229Google Scholar
7. Alciati A, Fusi A, D’Arminio Monforte A, et al: New-onset delusions and hallucinations in patients infected with HIV. J Psychiatry Neurosci 2001; 263:229–234Google Scholar
8. Isacson O, Bjorklund LM, Schumacher JM: Toward full restoration of synaptic and terminal function of the dopaminergic system in Parkinson’s disease by stem cells. Ann Neurol 2003; 53(suppl 3):S1135–1146Google Scholar
9. Lindvall O: Stem cells for cell therapy in Parkinson’s disease. Pharmacol Res 2003; 4 7 4:279–287 Google Scholar
10. McBride JL, Kordower JH: Neuroprotection for Parkinson’s disease using viral vector-mediated delivery of GDNF. Prog Brain Res 2002; 138:421–432Google Scholar
11. Hurelbrink CB, Barker RA: The potential of GDNF as a treatment for Parkinson’s disease. Exp Neurol 2004; 185:1–6Google Scholar
12. Jollivet C, Aubert-Pouessel A, Clavreul A, et al: Striatal implantation of GDNF releasing biodegradable microspheres promotes recovery of motor function in a partial model of Parkinson’s disease. Biomater 2004; 25:933–942Google Scholar
13. Sidhu A, Wersinger C, Vernier P: Alpha-Synuclein regulation of the dopaminergic transporter: a possible role in the pathogenesis of Parkinson’s disease. FEBS Lett 2004; 56:1–5Google Scholar
14. Spillantini MG, Crowther Ra, Jakes R, et al: Alpha-synuclein in filamentous inclusions of lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc Natl Acad Sci 1998; 95:6469–6473Google Scholar
15. Dickson DW: Tau and synuclein and their role in neuropathology. Brain Pathol 1999; 9:657–661Google Scholar
16. Trojanowski JQ, Lee VM: Aggregation of neurofilament and alpha-synuclein proteins in lewy bodies: implications for the pathogenesis of Parkinson disease and lewy body dementia. Arch Neurol 1998; 55:151–152Google Scholar
17. Harding AJ, Stimson E, Henderson JM, et al: Clinical correlates of selective pathology in the amygdala of patients with Parkinson’s disease. Brain 2002; 11:2431–2445Google Scholar
18. Lavedan C: The synuclein family. Genome Res 1998; 8:871–880Google Scholar
19. Clayton DF, George JM: The synucleins: a family of proteins involved in synaptic function, plasticity, neurodegeneration and disease. Trends Neurosci 1998; 21:249–254Google Scholar
20. Maroteaux L, Campanelli JT, Scheller RH: Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J Neurosci 1988; 8:2804–2815Google Scholar
21. Jenco JM, Rawlingson A, Daniels B, et al: Regulation of phospholipase D 2 : selective inhibition of mammalian phospholipase D isoenzymes by alpha- and beta-synucleins. Biochemistry 1998; 37:4901–4909 Google Scholar
22. Rochet JC, Outeiro TF, Conway KA, et al: Interactions among alpha-synuclein, dopamine, and biomembranes: some clues for understanding neurodegeneration in Parkinson’s disease. J Mol Neurosci 2004; 23:23–34Google Scholar
23. Polymeropoulos MH, Lavedan C, Leroy E, et al: Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997; 276:2045–2047Google Scholar
24. Papapetropoulos S, Paschalis C, Athanassiadou A, et al: Clinical phenotype in patients with alpha-synuclein Parkinson’s disease living in greece in comparison with patients with sporadic Parkinson’s disease. J Neurol Neurosurg Psychiatry 2001; 70:662–665Google Scholar
25. Farrer M, Kachergus J, Forno L: Comparison of kindreds with Parkinsonism and alpha-synuclein genomic multiplications. Ann Neurol 2004; 55:174–179Google Scholar
26. Betarbet R, Sherer TB, MacKenzie G, et al: Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat Neurosci 2000; 3:1301–1306Google Scholar
27. Feany MB, Bender WW: A drosophila model of Parkinson’s disease. Nature 2000; 404:394–398Google Scholar
28. Masliah E, Rockenstein E, Veinbergs I, et al: Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science 2000; 287:1265–1269Google Scholar
29. Uhl GR: Dopamine transporter: basic science and human variation of a key molecule for dopaminergic function, locomotion, and Parkinsonism. Mov Disord 2003; 18(suppl 7):S71–80Google Scholar
30. Braak H, Braak E, Yilmazer D, et al: Nigral and extranigral pathology in Parkinson’s disease. J Neural Transm Suppl 1995; 46:15–31Google Scholar
31. Jellinger KA: Pathology of Parkinson’s disease: changes other than the nigrostriatal pathway. Mol Chem Neuropathol 1991; 14:153–197Google Scholar
32. Braak H, Braak E: Pathoanatomy of Parkinson’s disease. J Neurol 2000; 247(suppl 2):3–10Google Scholar
33. Rub U, Del Tredici K, Schultz C, et al: Parkinson’s disease: the thalamic components of the limbic loop are severely impaired by alpha-synuclein immunopositive inclusion body pathology. Neurobiol Aging 2002; 23:245–254Google Scholar
34. Gomez-Tortosa E, Newell K, Irizarry MC, et al: Clinical and quantitative pathologic correlates of dementia with lewy bodies. Neurology 1999; 53:1284–1291Google Scholar
35. Fearnley JM, Lees AJ: Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain 1991; 114:2283–2301Google Scholar
36. Gibb WR, Lees AJ: Anatomy, pigmentation, ventral and dorsal subpopulations of the substantia nigra, and differential cell death in Parkinson’s disease. J Neurol Neurosurg Psychiatry 1991; 54:388–396Google Scholar
37. Galter D, Buervenich S, Carmine A, et al: Aldh1 mRNA: presence in human dopamine neurons and decreases in substantia nigra in Parkinson’s disease and in the ventral tegmental area in schizophrenia. Neurobiol Disord 2003; 14:637–647Google Scholar
38. Uhl GR, Hedreen JC, Price DL: Parkinson’s disease: loss of neurons from the ventral tegmental area contralateral to therapeutic surgical lesions. Neurology 1985; 35:1215–1218Google Scholar
39. Haber SN, McFarland NR: The concept of the ventral striatum in nonhuman primates. Acad Sci 1999; 877:33–48Google Scholar
40. Hirsch E, Graybiel AM, Agid YA: Melanized dopaminergic neurons are differentially susceptible to degeneration in Parkinson’s disease. Nature 1988; 334:345–348Google Scholar
41. Heimer L, Wilson RD: The subcortical projections of the allocortex: similarities in the neural associations of the hippocampus, the piriform cortex, and the neocortex, in Golgi Centennial Symposium. Edited by Santini M. New York, Raven Press, 1975Google Scholar
42. Koob GF, Nestler EJ: The neurobiology of drug addiction. J Neuropsychiatry Clin Neurosci 1997; 9:482–497Google Scholar
43. Heimer L: Basal forebrain in the context of schizophrenia. Brain Res 2000; 31(3):205–235Google Scholar
44. Ballmaier M, Zoli M, Leo G, et al: Preferential alterations in the mesolimbic dopamine pathway of heterozygous reeler mice: an emerging animal-based model of schizophrenia. Eur J Neurosci 2002; 15:1197–1205Google Scholar
45. O’Donnell P, Grace AA: Different effects of subchronic clozapine and haloperidol on dye-coupling between neurons in the rat striatal complex. Neuroscience 1995; 66:763–767Google Scholar
46. Young WS, Alheid GF, Heimer L: The ventral pallidal projection to the mediodorsal thalamus: a study with fluorescent retrograde tracers and immunohistofluorescence. J Neurosci 1984; 4:1626–1638Google Scholar
47. Lavin A, Grace AA: Modulation of dorsal thalamic cell activity by the ventral pallidum: its role in the regulation of thalamocortical activity by the basal ganglia. Synapse 1994; 18:104–127Google Scholar
48. Grace AA: Gating of information flow within the limbic system and the pathophysiology of schizophrenia. Brain Res 2000; 31:330–341Google Scholar
49. Rolls ET: Neurophysiology and cognitive functions of the striatum. Rev Neurol 1994; 150:648–660Google Scholar
50. Swerdlow NR, Geyer M: Using an animal model of deficient sensorimotor gating to study the pathophysiology and new treatments in schizophrenia. Schizophr Bull 1998; 20:671–684Google Scholar
51. Gloor P, Olivier A, Quesney LF: The role of the limbic system in experiential phenomena of temporal lobe epilepsy. Ann Neurol 1982; 12:129–144Google Scholar
52. Torrey EF, Peterson MR: Schizophrenia and the limbic system. Lancet 1974; 2(7886):942–946Google Scholar
53. Harding AJ, Broe GA, Halliday GM: Visual hallucinations in lewy body disease relate to lewy bodies in the temporal lobe. Brain 2002; 125:391–403Google Scholar
54. Yilmazer-Hanke DM, Wolf HK, Schramm J, et al: Subregional pathology of the amygdala complex and entorhinal region in surgical specimens from patients with pharmacoresistant temporal lobe epilepsy. J Neuropathol Exp Neurol 2000; 59:907–920Google Scholar
55. Bancaud J, Brunet-Bourgin F, Chauvel P: Anatomical origin of deja vu and vivid “memories” in human temporal lobe epilepsy. Brain 1994; 117:71–90Google Scholar
56. Anderson AK, Phelps EA: Intact recognition of vocal expressions of fear following bilateral lesions of the human amygdala. Neuroreport 1998; 9:3607–3613Google Scholar
57. Gray TS: Functional and anatomical relationships among the amygdala, basal forebrain, ventral striatum, and cortex: an integrative discussion. Ann NY Acad Sci 1999; 877:439–444Google Scholar
58. Morris JS, DeGelder B, Weiskrantz L, et al: Differential extrageniculostriate and amygdala responses to presentation of emotional faces in a cortically blind field. Brain 2001; 124:1241–1252Google Scholar
59. Rosenkranz JA, Grace AA: Cellular mechanisms of infralimbic and prelimbic prefrontal cortical inhibition and dopaminergic modulation of basolateral amygdala neurons in vivo. J Neurosci 2002; 22:324–337Google Scholar
60. Carlsson A, Waters N, Holm-Waters S, et al: Interactions between monoamines, glutamate, and gaba in schizophrenia: new evidence. Annu Rev Pharmacol Toxicol 2001; 41:237–260Google Scholar
61. Farmer AE , McGuffin P: The pathogenesis and management of schizophrenia. Drugs 1988; 35:177–185Google Scholar
62. Lieberman A: Managing the neuropsychiatric symptoms of Parkinson’s disease. Neurology 1998; 50(suppl 6):S33–38Google Scholar
63. Wolters EC: Dopaminomimetic psychosis in Parkinson’s disease patients: diagnosis and treatment. Neurology 1999; 52:10–13Google Scholar
64. Juncos JL, Roberts VJ, Evatt ML, et al: Quetiapine improves psychotic symptoms and cognition in Parkinson’s disease. Mov Disord 2004; 19:29–35Google Scholar
65. Torres GE, Gainetdinov RR, Caron MG: Plasma membrane monoamine transporters: structure, regulation and function. Nat Rev Neurosci 2003; 4:13–25Google Scholar
66. Giros B, Caron MG: Molecular characterization of the dopamine transporter. Trends Pharmacol Sci 1993;14:43–49Google Scholar
67. Haber SN, Ryoo H, Cox C: Subsets of midbrain dopaminergic neurons in monkeys are distinguished by different levels of mRNA for the dopamine transporter: comparison with the mrna for the D 2 receptor, tyrosine hydroxylase and calbindin immunoreactivity. J Comp Neurol 1995; 362:400–410 Google Scholar
68. Wilson JM, Nobrega JN, Carroll ME, et al: Heterogeneous subregional binding patterns of 3H-WIN 35,428 and 3H-GBR 12,935 are differentially regulated by chronic cocaine self-administration. J Neurosci 1994; 14:2966–2979Google Scholar
69. Gordon I, Weizman R, Rehavi M: Modulatory effect of agents active in the presynaptic dopaminergic system on 74he striatal dopamine transporter. Eur J Pharmacol 1996; 298:27–30Google Scholar
70. Moody CA, Granneman JG, Bannon MJ: Dopamine transporter binding in rat striatum and nucleus accumbens is unaltered following chronic changes in dopamine levels. Neurosci Lett 1996; 21:55–57Google Scholar
71. Vander Borght T, Kilbourn M, Desmond T, et al: The vesicular monoamine transporter is not regulated by dopaminergic drug treatments. Eur J Pharmacol 1995; 294:577–583Google Scholar
72. Murer MG, Dziewczapolski G, Menalled LB, et al: Chronic levodopa is not toxic for remaining dopamine neurons, but instead promotes their recovery, in rats with moderate nigrostriatal lesions. Ann Neurol 1998; 43:561–575Google Scholar
73. Innis RB, Marek KL, Sheff K: Effect of treatment with L-dopa/carbidopa or L-selegiline on striatal dopamine transporter spect imaging with [123I]beta-CIT. Mov Disord 1999; 14:436–442Google Scholar
74. Ahlskog JE, Uitti RJ, O’Connor MK, et al: The effect of dopamine agonist therapy on dopamine transporter imaging in Parkinson’s disease. Mov Disord 1999; 14:940–946Google Scholar
75. Nurmi E, Bergman J, Eskola O, et al: Reproducibility and effect of levodopa on dopamine transporter function measurements: a [18F]CFT PET study. J Cereb Blood Flow Metab 2000; 20:1604–1609Google Scholar
76. Ikawa K, Watanabe A, Kaneno S, et al: Modulation of [3H]mazindol binding sites in rat striatum by dopaminergic agents. Eur J Pharmacol 1993; 250:261–266Google Scholar
77. Gnanalingham KK, Robertson RG: The effects of chronic continuous versus intermittent levodopa treatments on striatal and extrastriatal D 1 and D 2 dopamine receptors and dopamine uptake sites in the 6-hydroxydopamine lesioned rat—an autoradiographic study. Brain Res 1994; 640:185–194 Google Scholar
78. Boja JW, Vaugh R, Patel A, et al: The Dopamine Transporter, in Dopamine Receptors and Transporter: Pharmacology, Structure and Function. Edited by Niznik HB. 1994Google Scholar
79. Engber TM, Susel Z, Kuo S, et al: Levodopa replacement therapy alters enzyme activities in striatum and neuropeptide content in striatal output regions of 6-hydroxydopamine lesioned rats. Brain Res 1991; 552:113–118Google Scholar
80. Joyce JN, Meador-Woodruff JH: Linking the family of D 2 receptors to neuronal circuits in human brain: insights into schizophrenia. Neuropsychopharmacology 1997; 16:375–384 Google Scholar
81. Alexander GE, Crutcher MD, DeLong MR: Basal ganglia-thalamocortical circuits: parallel substrates for motor, oculomotor, “prefrontal” and “limbic” functions. Prog Brain Res 1990; 85:119–146Google Scholar
82. Lu XY, Ghasemzadeh MB, Kalivas PW: Expression of D 1 receptor, D 2 receptor, substance P and enkephalin messenger rnas in the neurons projecting from the nucleus accumbens. Neuroscience 1998; 82:767–780 Google Scholar
83. Curran EJ, Watson SJ: Dopamine receptor mrna expression patterns by opioid peptide cells in the nucleus accumbens of the rat: a double in situ hybridization study. J Comp Neurol 1995; 361:57–76Google Scholar
84. Betarbet R, Greenamyre JT: Regulation of dopamine receptor and neuropeptide expression in the basal ganglia of monkeys treated with MPTP. Exp Neurol 2004; 189:393–403Google Scholar
85. Frohna PA, Rothblat DS, Joyce JN, et al: Alterations in dopamine uptake sites and D 1 and D 2 receptors in cats symptomatic for and recovered from experimental Parkinsonism. Synapse 1995; 19:46–55 Google Scholar
86. Gnanalingham KK, Smith LA, Hunter AJ, et al: Alterations in striatal and extrastriatal D-1 and D-2 dopamine receptors in the MPTP-treated common marmoset: an autoradiographic study. Synapse 1993; 14:184–194Google Scholar
87. Piggott MA, Marshall EF, Thomas N, et al: Striatal dopaminergic markers in dementia with lewy bodies, Alzheimer’s and Parkinson’s diseases: rostrocaudal distribution. Brain 1999; 122:1449–1468Google Scholar
88. Pope-Coleman A, Tinker JP, Schneider JS: Effects of GM1 ganglioside treatment on pre- and postsynaptic dopaminergic markers in the striatum of Parkinsonian monkeys. Synapse 2000; 36:120–128Google Scholar
89. Rioux L, Frohna PA, Joyce JN, et al: The effects of chronic levodopa treatment on pre- and postsynaptic markers of dopaminergic function in striatum of Parkinsonian monkeys. Mov Disord 1997; 12:148–158Google Scholar
90. Rinne JO, Laihinen A, Ruottinen H, et al: Increased density of dopamine D 2 receptors in the putamen, but not in the caudate nucleus in early Parkinson’s disease: a PET study with [11C]raclopride. J Neurol Sci 1995; 132:156–161 Google Scholar
91. Graham WC, Clarke CE, Boyce S, et al: Autoradiographic studies in animal models of hemi-Parkinsonism reveal dopamine D 2 but not D 1 receptor supersensitivity, II: Unilateral intra-carotid infusion of MPTP in the monkey ( Macaca fascicularis). Brain Res 1990; 514:103–110 Google Scholar
92. Przedborski S, Jackson-Lewis V, Popilskis S, et al: Unilateral MPTP-induced Parkinsonism in monkeys: a quantitative autoradiographic study of dopamine D 1 and D 2 receptors and re-uptake sites. Neurochirurgie 1991; 37:377–382 Google Scholar
93. Gaser C, Nenadic I, Volz HP, et al: Neuroanatomy of “hearing voices”: a frontotemporal brain structural abnormality associated with auditory hallucinations in schizophrenia. Cereb Cortex 2004; 14:91–96Google Scholar
94. Lennox BR, SB Park, Medley I, et al: The functional anatomy of auditory hallucinations in schizophrenia. Psychiatry Res 2000; 100:13–20Google Scholar
95. Silbersweig DA, Stern E, Frith C, et al: A functional neuroanatomy of hallucinations in schizophrenia. Nature 1995; 378:76–79Google Scholar

