
Huntington's Disease: Effect of Memantine on FDG-PET Brain Metabolism?
Abstract
In this open-label pilot study, the authors evaluated the effect of memantine on the distribution of brain glucose metabolism in four Huntington's disease (HD) patients as determined by serial 18-fluoro-deoxyglucose [F18]FDG-PET scans over a period of 3–4 months (90–129 days, with one patient choosing to continue treatment over the 18-month follow-up period). The treatment regimen was well tolerated. No significant differences on neuropsychological parameters before and after treatment were detected; but the patient who continued treatment did not deteriorate at 18 months' reevaluation, whereas the three patients who had stopped treatment after 3 to 4 months had minor progression in all cognitive domains on re-evaluation 12 months after the end of treatment.
Huntington's disease (HD) is an autosomal dominantly-inherited progressive neurodegenerative disorder caused by a CAG triplet repeat-expansion in the HTT gene. Symptoms include chorea, psychiatric disturbances, and cognitive decline. Several potential therapies are assessed in human clinical trials, but no treatment significantly alters the inexorable progression of this disorder. Furthermore, no objective biochemical measures to follow the progression of HD are yet available, and the search to identify biomarkers, essential for rapid and reliable evaluation in clinical trials, is urgent.1 For unknown reasons, GABA-ergic medium spiny striatal neurons (MSN) are especially sensitive to the neurodegeneration of HD. Dysregulation of glutamate/calcium-signaling pathways may be one potential cause,2 which suggests that antagonists of glutamate-signaling pathways may have a beneficial value for treatment of HD patients. Memantine is a specific, uncompetitive, open-channel NMDA-receptor antagonist, stabilizing glutamatergic tone and, in an in vitro assay with cultured MSN from YAC128 transgenic mice, memantine was found to exert significant neuroprotection.3 Clinical efficacy of memantine has been evaluated in two open-label studies. One study, with 27 HD patients followed for 2 years, suggested that memantine treatment might decrease progression of the symptoms.4 In another study, with nine patients followed for 3 months, memantine was found to improve motor functioning.5 In this open-label pilot study, the authors decided to evaluate the effect of memantine on the distribution of brain glucose metabolism in HD patients as determined by serial 18-fluoro-deoxyglucose [F18]FDG-PET scans. This technique has proven to be sensitive enough to diagnose neurodegenerative diseases; also, changes in FDG uptake can be manifest before overt clinical or morphological changes.6,7
METHOD
Colleagues donated their yearly, free, one-sample of memantine tablets for this study, a quantity allowing us to treat four HD patients for approximately 3 to 4 months. After they gave informed consent, four consecutive patients with clinical and molecular-genetically-verified HD (ages 42 to 65 years) were enrolled in an open-label study with memantine, titrated from 5 mg to 20 mg/day in 2 divided doses over 4 weeks. Treatment was continued for 90 days to 129 days. Because of a subjective impression of stabilization, one patient chose to continue medication and pay for it, and he was also reevaluated after 18 months, still on treatment (Figure 1). Other medications were unchanged during this period (Table 1). We administered the Unified Huntington's Disease Rating Scale (UHDRS) Motor score, Total Functional Capacity Score (TFC), Mini-Mental State Exam (MMSE), Frontal Assessment Battery (FAB), Epworth Sleepiness Scale, and a comprehensive neuropsychological battery. The test battery comprised the following factors: General Intellectual Functioning: Vocabulary and Information from the WAIS (Wechsler Adult Intelligence Scale); Learning and Memory: Selective Reminding Test and Rey Complex Figure Test (3-minute recall); Psychomotor Speed: Trail-Making Test part A and Symbol Digit Modalities Test (SDMT); Attention and Executive Functions: Digit Span Forward and Backward, Trail-Making Test part B, Stroop Interference Test, Wisconsin Card-Sorting Test, Verbal Fluency (“S” words and Animals in 1 minute), continuous subtraction 100–7; Visuospatial Functions: Rey Complex Figure Test (copy) and Block Design (WAIS); Abstract Reasoning: Similarities (WAIS), Picture Arrangement (WAIS), Raven's Progressive Matrices Advanced (Set 1), Picture Completion (WAIS–III), and Matrices (WAIS–III). Test performances were compared with age-adjusted normative data. All tests were performed 1 week before initiating treatment and again 1 week before ending treatment, and, in Patient #4, again after 18 months of the extended treatment period.
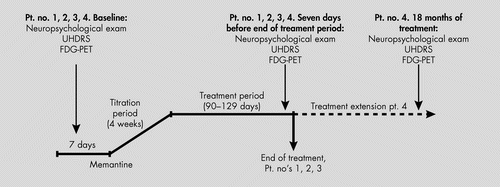
UHDRS: Unified Huntington's Disease Rating Scale.
 |
[18F]FDG-PET scans were accordingly performed before and at the end of the treatment period (90 days to 129 days). One patient, who continued treatment, had a third [18F]FDG-PET scan after 18 months. Eleven healthy, nonmedicated volunteers (8 men, 3 women), with a median age of 26 years (age 23–50 years) and no history of neurological or psychiatric disorders were [18F]FDG-PET–scanned as a control group.
PET Scanning
We performed PET scans with an 18-ring GE-Advance scanner (General Electric Medical Systems; Milwaukee, WI, U.S.) operating in 3D-acquisition mode, producing 35 image slices, with an interslice distance of 4.25 mm. The total axial field of view was 15.2 cm., with an approximate in-plane resolution of 5 mm. Before the scans were taken, an intravenous bolus of approximately 200 MBq FDG was injected in the left antecubital vein while the subject rested in supine position with eyes covered and noise level kept at a minimum to keep alertness at the lowest level; 30 minutes later, the subject was positioned in the scanner, with his or her head fixed, to restrict movements. A 10-minute transmission scan was performed for attenuation-correction, followed by a 10-minute 3D emission scan. PET images of the FDG distribution were reconstructed, using a 4.0-mm. Hanning filter transaxially and an 8.5-mm. Ramp filter axially for routine clinical evaluation. The reconstruction matrix was 128×128 in-plane, with a 2×2×4.25-mm. voxel dimension. All scans were performed in the morning.
Image Analysis
The images were analyzed by use of two strategies: regions of interest (ROI) and voxel-based.
All PET intrasubject images were aligned to the first scan by using the “True-D” program in the Siemens Multimodality Workplace for the ROI-based analyses. Circular, 1-cm-diameter ROIs were placed over the caudate nuclei and the thalami, symmetrically and bilaterally, and a large, irregular ROI covering frontal, parietal, temporal, and occipital cortical gray matter. Identical ROIs were used to follow changes in time. The ROI values of the two thalami and the two striata were averaged and normalized to the values of the cortical ROIs. An identical strategy was used for the healthy-control subjects. The ROI values were evaluated with a paired t-test, evaluating the effect of the treatment, and an unpaired t-test, evaluating differences between HD patients before treatment and controls, with a significance threshold of p<0.05. For the voxel-based image analysis, the authors used statistical parametric mapping software (SPM5; Wellcome; Department of Cognitive Neurology; London, UK (http://www.fil.ion.ucl.ac.uk/spm/). The complete PET brain volume was sampled in all subjects. Intra-subject PET images were aligned with a 3D, six-parameter rigid body transformation, and transformed into the standard stereotaxic atlas of Talairach and Tournoux,8 using the PET template from MNI (Montreal Neurological Institute). PET images were smoothed with a 16-mm. FWHM isotropic Gaussian kernel, and differences in global activity were removed by ratio adjustment, estimating global activity independently of changes in local activity. Statistical analyses were performed using unpaired t-tests of patients versus controls in order to document the extent of disease at the time of treatment initiation. Also, paired t-tests were used in the patient group to identify significant treatment effects in the first treatment period. Statistical thresholds were set to p<0.05, FDR (false discovery rate)-corrected9 for non-independent comparisons at the voxel level for clusters and an extent threshold above 200 voxels. The approximate anatomical localizations of significant results were subsequently obtained from a communal normalized MRI image averaged across all subjects.
RESULTS
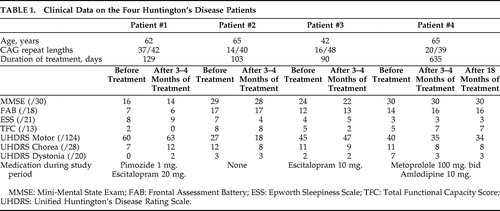
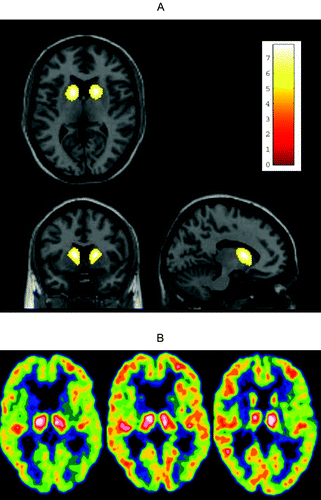
The treatment regimen was well tolerated. No significant differences on neuropsychological parameters before and after treatment were detected; but Patient #4, who continued treatment, did not deteriorate at 18 months' reevaluation, whereas the three patients who had stopped treatment after 3 to 4 months had minor progression in all cognitive domains on re-evaluation 12 months after the end of treatment. The UHDRS Motor score improved from 27 to 18 in one patient (Patient #2), but was mainly unchanged in the other patients; however, with a decrease from 40 to 35, and 34 after 3 and 18 months of treatment, respectively, in Patient #4, who continued treatment (Table 1). Treatment reduced the average thalamic glucose metabolism relative to the cortical values from 1.32 (standard deviation [SD]: 0.08) to 1.22 (SD: 0.05; p=0.05). A decrease in the thalami/cortex ratio was seen in all patients. However, the p value was not significant by our selected criteria. Also, this ratio was not significantly different from healthy-controls; mean ratio: 1.21 (SD: 0.11; p=0.11). The corresponding average glucose metabolism relative to the cortical values for the striata was 0.71 (SD: 0.06) and 0.71 (SD: 0.09; Table 1). Compared with healthy-controls, this was significantly reduced: mean ratio: 1.23 (SD: 0.12; p<0.0001. These results were corroborated by the voxel-level analysis, revealing a significant reduction of uptake bilaterally in the anterior section of the striatum before the treatment (Figure 2 [A]). There were no other areas of significant changes. Treatment did not improve cortical or striatal metabolism, as compared with the thalamus. On the other hand, the metabolism did not decrease during treatment, including after the prolonged treatment period in one patient, and a relative improvement in cortical metabolism as compared with thalamus was visually indicated in all four patients (Figure 2 [B]).
A: Statistical voxel-based analysis of [18F]FDG-PET scans in HD patients at baseline compared to a healthy control group. Significant areas of relatively reduced glucose metabolism displayed in t-values are showed in a “hot-metal” scale and thresholded at a level of p 5.5, after correction for multiple comparisons. There is a significant reduction in the anterior section of the striatum, and no other areas.
B: [18F]FDG-PET scans showing the changes in Patient 2 before treatment (left) and during treatment, after about 100 days (middle) compared to a Control (right). The thalami-to-cortex ratio in the 3 images were 1.38, 1.21, 1.22, respectively. The images are scaled to the activity in the thalami using a “Spectrum 10” color scale, where each discrete color isocontour corresponds to 1/10 of the extension of the scale. There is an impression of a relatively increased uptake in the cortex versus the thalami with treatment.
DISCUSSION
Treatment of HD, as well as reliable and reproduceable objective measurements to define progression of this devastating and heterogeneous disease, have been sought for years. In this trial, we united experience from earlier studies suggesting clinical effects of memantine on HD4,5 and [18F]FDG-PET-scans as a sensitive marker of disease-progression in other neurodegenerative disorders.6,7 Treatment of four HD patients with memantine 20 mg/day for 3 months–4 months revealed no significant changes in the clinical and neuropsychological parameters. The patient with prolonged treatment did not deteriorate after 18 months, whereas the three patients treated for only 3–4 months all had progression at neuropsychological examination after 12 months. The relative FDG distribution, using PET, is an indirect marker of regional synaptic activity,10 commonly used in the investigation of neurodegenerative diseases11,12 and in earlier studies, a decreased metabolism is described before clinical changes, using [18F]FDG-PET in pre-symptomatic mutation-positive persons.6 We did not quantify the regional cerebral glucose metabolism in absolute physiological units; this would have involved repeated arterial cannulations, and it would have been an unacceptable trauma to most patients. Thus, in this investigation we can only observe changes in regional cerebral glucose metabolism relative to healthy-control subjects, and relative to other cerebral regions. Our data might represent a potential positive effect on the damaged synaptic transmission in the basal ganglia, as the cortical and striatal metabolism as assessed by [18F]FDG-PET was unchanged during the treatment period even in the patient who continued treatment and was scanned a third time after 18 months. Because this study did not include an untreated group of matched patients, we cannot estimate the natural cause of the relative regional cerebral glucose metabolism and can only hypothesize about the findings. From the literature, we would expect an average reduction per year of 2.3%–3.1% in caudate glucose metabolic rate.13,14 Before treatment, there was relatively high thalamic activity relative to the cortex, which was reduced with treatment, to approach the value in the control group. This could be caused by either a reduction in thalamic metabolism or an increase in cortical metabolism. If the latter were the case, this would imply a relative increase in striatae; however, these statements are hypothetical interpretations of the trends in the data. This pilot study implies further limitations in the interpretation of the results: The hypothesized disease-modifying effect of memantine may be correct, but an acute pharmacological effect is another possibility. Furthermore, this study is too small to illustrate or exclude a tentative modifier effect of the patients' genotype besides the possible drug effect. Finally, a nontreated control group of matched HD patients would have been preferable. Treatment options are desperately needed, especially those with disease-modifying potential. This study can only provide preliminary data, and a long-term, randomized, double-blinded study design including [18F]FDG-PET scans is highly desirable to further explore a potential positive effect of memantine.
1. : Biomarkers for neurodegenerative diseases. Curr Opin Neurol 2005; 18:698–705Crossref, Medline, Google Scholar
2. : Inositol 1,4,5-tripshosphate receptor, calcium signalling, and Huntington's disease. Subcell Biochem 2007; 45:323–335Crossref, Medline, Google Scholar
3. : Evaluation of clinically relevant glutamate pathway inhibitors in in-vitro model of Huntington's disease. Neurosci Lett 2006; 407:219–223Crossref, Medline, Google Scholar
4. : The N-methyl-D-aspartate antagonist memantine retards progression of Huntington's disease. J Neural Transm Suppl 2004; 68:117–122Crossref, Medline, Google Scholar
5. : A pilot study of the clinical efficacy and safety of memantine for Huntington's disease. Parkinson Relat Disord 2007; 13:453–454Crossref, Medline, Google Scholar
6. : Thalamic metabolism and symptom onset in preclinical Huntington's disease. Brain 2007; 130:2858–2867Crossref, Medline, Google Scholar
7. : Brain white-matter volume loss and glucose hypometabolism precede the clinical symptoms of Huntington's disease. J Nucl Med 2006; 47:215–222Medline, Google Scholar
8. : Co-planar stereotaxic atlas of the human brain. New York, Thieme Medical Inc., 1988Google Scholar
9. : Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc B 1995; 57:289–300Google Scholar
10. : Circulatory and metabolic correlates of brain function in normal humans, in: Handbook of Physiology: The Nervous System. Edited by Plum FMV. Baltimore, MD, Williams & Wilkins, 1987, pp 643–674Google Scholar
11. : Brain glucose metabolism in the early and specific diagnosis of Alzheimer's disease: FDG-PET studies in MCI and AD. Eur J Nucl Med Mol Imag 2005; 32:486–510Crossref, Medline, Google Scholar
12. : Brain 18F-FDG PET in the diagnosis of neurodegenerative dementias: comparison with perfusion SPECT and with clinical evaluations lacking nuclear imaging. J Nucl Med 2004; 45:594–607Medline, Google Scholar
13. : Serial changes of cerebral glucose metabolism and caudate size in persons at risk for Huntington's disease. Arch Neurol 1992; 49:1161–1167Crossref, Medline, Google Scholar
14. : Striatal glucose metabolism and dopamine D2-receptor binding in asymptomatic gene carriers and patients with Huntington's disease. Brain 1996; 119:2085–2095Crossref, Medline, Google Scholar

