
Post-Traumatic Epilepsy: Review of Risks, Pathophysiology, and Potential Biomarkers
Abstract
Epilepsy is a disorder of the brain characterized by an enduring predisposition to generate epileptic seizures. An epileptic seizure is a transient occurrence of signs and/or symptoms because of abnormal excessive or synchronous neuronal activity in the brain.6 The definition of epilepsy requires the occurrence of at least one unprovoked epileptic seizure. Post-traumatic epilepsy (PTE) is a recurrent seizure disorder secondary to traumatic brain injury (TBI).7–10 Five percent of all epilepsy and 20% of structural epilepsy in the general population is PTE.7 PTE is also the most common cause of new-onset epilepsy in young adults.11
Structural epilepsies typically develop in three phases: a brain insult (e.g., TBI, stroke, infections, tumor) leading to epileptogenesis (latency period, prior to the onset of seizures), followed by recurrent unprovoked seizures (epilepsy). Seizures that occur early versus late after TBI have different implications for prognosis and management.7–10 Seizures during the acute phase (within the first week after trauma) have a low likelihood of recurrence, whereas later seizures are likely to recur and so may represent epilepsy. Early seizures appear to result from the direct physical injury. These are acute symptomatic events and are not believed to represent epilepsy. Immediate seizures are a distinct category of early seizures, those occurring within 24 hours of trauma. These are thought to represent “convulsive concussions” because of brief traumatic functional decerebration that results from cortical inhibition and not epileptic events.12 The risk of PTE is highest within the first 2 years following a brain injury with most patients who have a late post-traumatic seizure experiencing a second seizure within 2 years.13 A high seizure frequency in the first year of onset predicts future seizure severity and medical intractability.14 However, a substantial minority (15%‒20%) of patients may experience their first seizure beyond 2 years (Figure 1).7
Ictal semiology, the signs and symptoms that are produced from an epileptic seizure, differ depending on the seizure type and location (Figure 1). The clinical manifestations are extremely variable and depend on the cortical areas involved. Patients with frontal lobe epilepsy may exhibit semi-purposeful complex motor behaviors such as kicking, screaming, and thrashing episodes. These seizures can often be mistaken for psychogenic nonepileptic events. Clinically, frontal lobe seizures differ from nonepileptic events in that they are more stereotyped, nongoal directed, and shorter in duration.15 Partial seizures of temporal lobe origin may also present with emotional symptoms such as fear or panic, followed by periods of postictal confusion and amnesia.
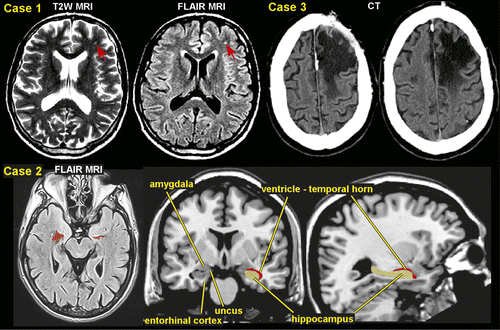
There have been several observational studies looking at the type of seizures that develop following moderate-severe TBI, but no consistent patterns have emerged.13,16,17 In up to two-thirds of patients, late post-traumatic seizures are generalized or focal with secondary generalization, and often both seizure types may coexist.13,17–20 Retrospective studies of adult patients with medically intractable epilepsy have reported focal onset seizures in 78%‒91%: temporal lobe in 15%‒56%, frontal lobe in 23%‒36%, occipital lobe in 2%‒6%, and parietal lobe in 5%‒38%.20,21 One study reported that parietal semiology was observed more frequently in patients with penetrating trauma, whereas patients with blunt trauma showed higher temporal and frontal semiologies.20 Although an earlier study indicated that PTE manifested as medial temporal lobe epilepsy (TLE) only in patients injured at or before age 5, later studies have confirmed (based on seizure semiology, electrophysiology, and presence of mesial temporal sclerosis) medial TLE in 30%‒35% of patients injured at older ages.16,21,22 Hippocampal degeneration has been shown to be more severe in patients surviving more than 6 months compared with patients surviving less than 1 week, suggesting injury processes can be progressive.23 Hippocampal atrophy is a common finding among chronic TBI survivors and is also seen in various experimental models of TBI (Figure 1).24 This has been found to be bilateral in many cases. Certain areas of the hippocampus are more vulnerable than others.25 Histopathological examination in a series of temporal lobectomies found that neocortical gliosis was present in all specimens and hippocampal neuronal loss occurred in 94% of the cases, confirming that trauma is able to induce hippocampal epilepsy without any other major risk factors.26
Risk Factors and Epidemiology
The risk factors for PTE include advanced age, penetrating injuries, injury severity (e.g. neurosurgical procedure, intracranial hemorrhage, greater than 5 mm midline shift, duration of coma >24 hours, loss of consciousness >24 hours, prolonged length of post-traumatic amnesia), biparietal or multiple contusions, and frontal or temporal locations of the lesion.7–10,27,28 In penetrating brain injury, a foreign body pierces the bony skull and passes into (and in some cases through) the substance of the brain. This leads to physical disruption of neurons, glial cells and fiber tracts. The estimated relative risk of seizures after penetrating war injuries is very high as compared with the risk in the general population.8,10 Studies of the consequences of missile injuries to the head from World War I up through conflicts in the Middle East during the 1980s have shown a remarkably consistent pattern in terms of the development of epilepsy after this severe form of TBI. The Vietnam Head Injury Study, for example, found that incidence of PTE was 53% during the 15 years after the injury, with half still having seizures after 15 years.29–31
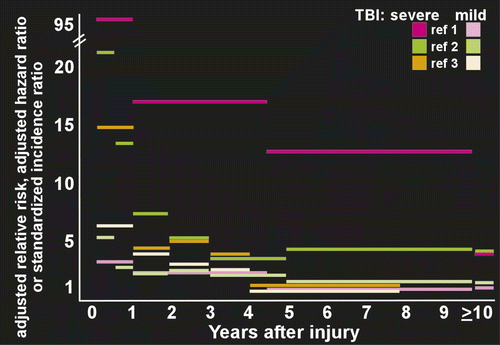
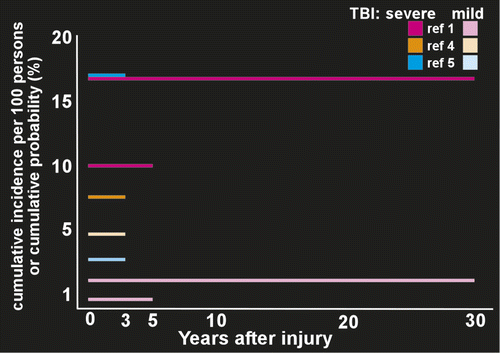
Several population based studies have measured risk of developing PTE in groups (most or all civilians with nonpenetrating injuries) containing both inpatients and outpatients (Figure 2). A study from Denmark (1,605,216 subjects; 78,572 had at least one TBI) found that compared with no brain injury, the relative risk of developing epilepsy for people older than 15 years was 3.5 after mild TBI and 12.2 after severe TBI.2 Risk was still elevated at 10 years after injury (1.5 for mild, 4.3 for severe). A U.S. study (4,541 subjects with a single TBI) reported 5-year cumulative PTE probability was 0.7% for mild, 1.2% for moderate, and 10% for severe TBI.1 The 30-year cumulative PTE probability was 2.1% for mild, 4.2% for moderate, and 16.7% for severe TBI. A study in Taiwan (559,658 subjects age 15 years or older; 19,336 had at least one TBI) found that compared with no brain injury the hazard ratio (a different measure of relative risk) for PTE was 3.9 for mild TBI and 7.8 for severe TBI.3 Population-based studies that focused on development of PTE following hospitalization have reported relatively similar results (Figure 2). A U.S. study (2,118 patients with TBI age 15 years or older) reported 3 years cumulative incidence per 100 persons of PTE was 9.1 across injury severity.4 The cumulative incidence per 100 persons was 4.4 for mild TBI, 7.6 for moderate TBI, and 13.6 for severe TBI. Peak onset was in year two.4 A study from China (2,826 patients with TBI age 4 to 79 years) reported a 3 year cumulative incidence of PTE of 5% across injury severity.5 Cumulative incidence was 3.6% for mild, 6.9% for moderate and 17%, for severe TBI. Onset was within the first year in the majority of cases.5 Although there are many methodological differences across these studies, these results suggest that even mild TBI is associated with clearly increased risk for PTE. Variability in seizure occurrence may also be attributable to the unique genetic makeup that each patient brings to post-TBI recovery as not every patient with clinical risk factors develops PTE.32–35
Epileptogenesis
The development of PTE after a latent period is an example of human epileptogensis, whereby a nonepileptic brain undergoes molecular and cellular alterations after a brain insult, which increases its excitability.7–9 This eventually leads to the occurrence of recurrent spontaneous seizures. Penetrating brain injury produces a cicatrix (scar) in the cortex and is associated with an increased risk of PTE of approximately 50%.29 The initial disruption of tissue may be compounded by ischemia and hemorrhage. Nonpenetrating injuries, including focal contusions and intracranial hemorrhages, are associated with a risk of PTE of up to 30%. In this setting, the mechanism of epileptogenesis may be partly related to the toxic effects of hemoglobin breakdown products on neuronal function.8,36 Closed head injuries produce diffuse axonal injury with shearing of axons, diffuse edema, and ischemia. The most common locations are the gray matter-white matter junction particularly in the frontal and temporal areas.37 This process leads to the release of excitatory amino acids, cytokines, bioactive lipids, and other toxic mediators causing secondary cellular injury.7–9 Epileptogensis is thought to be initiated by specific types of cell loss and neuronal reorganization, which results not only in enhanced excitation, but also in decreased inhibition, predisposing to hypersynchronization.38 Brain injuries also lead to upregulation of proinflammatory cytokines and activated immune responses to further increase seizure susceptibility, promote neuronal excitability, and impair blood–brain barrier (BBB) integrity.39,40 Early changes include the induction of immediate early genes and post-translational modifications of neurotransmitter receptor and ion channel/transporter proteins.41 Within days, neuronal death, initiation of an inflammatory cascade, and new gene transcription has been reported to occur.42 Later (days to weeks) anatomic changes include axonal sprouting and dendritic modifications. For example, mossy fiber sprouting can be observed in chronic epileptic brain.43 Over time, seizure threshold is lowered by a growing increase in excitability, and the risk of a seizure increases.44
The process by which trauma to the brain tissue leads to hyperexcitability, hypersynchronization, and development of recurrent seizures is relatively unknown, but there are several theories as to potential mechanisms based on experimental data, primarily from animal models of PTE. Animal models of TBI (mostly performed in rodents) are divided into focal brain injury, diffuse brain injury, mixed models of focal and diffuse brain injury, and models of repetitive concussive injury.45,46 These models utilize a direct impact on the epidural space on the brain tissue.45 A large number of studies have used lateral fluid-percussion or central fluid-percussion TBI models that produce both gray matter and white matter injury.47 Late spontaneous seizures (PTE) have consistently been reported in the rat fluid-percussion TBI model, which is the most commonly used model of human closed head TBI.47 The physiological effects from TBI are considered to consist of several phases including primary injury, evolution of primary injury, secondary injury, and regeneration.45,47 Multiple mechanisms are likely activated simultaneously or sequentially by brain trauma. Animal studies demonstrate that a large number of molecular and cellular alterations occur during the latent period that could be related to epileptogenesis, including cell death, gliosis, neurogenesis, axonal and dendritic plasticity, rearrangement of the extracellular matrix, and angiogenesis.47
Neuroimaging
Conventional neuroimaging methods including CT and MRI provide limited prognostic information for predicting development of PTE. Hippocampal atrophy associated with PTE is one of the most identified findings (Figure 1). There is promise with newer advanced MRI sequences that are more sensitive to microhemorrhages (e.g., susceptibility weighted imaging, SWI) and white matter injury (diffusion tensor imaging, DTI).48 The search for neuroimaging features that correlate with presence of PTE and so might serve as biomarkers indicating increased risk of developing PTE and/or of epileptogenic processes is still in the early stages.49,50
DTI shows potential for being more sensitive to the microstructural changes (e.g., gliosis, cell loss, axonal sprouting) thought to be involved in epileptogenesis than conventional MRI, as several studies have reported altered DTI metrics (lower fractional anisotropy [FA], higher mean diffusivity [MD]) in areas appearing normal on conventional MRI.51–55 One study found that FA was lower and the volume of abnormal tissue was greater in patients with TBI who developed PTE than in patients with TBI who did not.51 The authors noted that their results suggest that DTI may have the potential to predict presence of epileptogenesis following TBI.
As noted above, trauma-induced hemorrhage is associated with higher risk for PTE, and animal models suggest multiple potentially seizure-promoting changes including generation of substances that can induce further injury, such as highly reactive free radical oxidants.56 A longitudinal prospective study of patients with TBI (mild N=38, moderate N=26, severe N=71) used neuroimaging at multiple time after injury to assess whether the presence of gliosis and/or residual hemorrhage (hemosiderin) during the first 2 years increased risk of developing PTE.57 As expected, patients with focal brain lesions requiring surgical treatment had the highest risk of developing PTE. Neuroimaging evidence of contusion in which residual hemosiderin was only partially encompassed by gliosis at earlier times after injury, particularly those that progressed to full enclosure at later times, was also associated with a higher risk of PTE. Presence of small areas of hemosiderin with no associated gliosis was not associated with higher risk of PTE. No evidence of medial temporal sclerosis was present. As noted by the authors of this study, this might be due to the relatively early phase (up to two years after injury) and/or the lower resolution of the neuroimaging (1.0 tesla, 5mm slice thickness).
Focal alterations in BBB permeability are common following TBI, with biphasic changes reported in animal models.58,59 The early (hours) increase in permeability is likely a direct result of the injuring event, whereas the delayed (days) increase in permeability probably reflects secondary events. A study of patients with mild TBI (median 3 months post-injury) with (N=19) and without (N=18) PTE reported that BBB disruption (identified by contrast-enhanced MRI) was present more frequently in the PTE group (82.4% versus 25%) and the volume of cortex affected was larger.60 BBB dysfunction is seizure-promoting and may contribute to epileptogenesis.39,58,59 Activation of astrocytes has been reported following BBB disruption, perhaps a result of entry into the brain parenchyma of injurious serum constitutes (e.g. albumin), and is associated with epileptogenesis in animal models.39,58,59
Conclusions
Traumatic brain injury is a heterogeneous disorder involving several different mechanisms including concussive forces, acceleration-deceleration forces, blast injury, and projectile missiles. Each of these mechanisms can result in numerous clinical sequelae including but not limited to PTE. Numerous studies have documented the risks associated with the development of PTE with moderate to severe brain injuries conferring the greatest risk. The characteristics of the seizure itself will also reflect the anatomic heterogeneity of TBI, with the majority of the seizure foci occurring in the temporal and frontal lobes. The process of epileptogenesis that occurs as a result of a brain injury is still not fully understood but it remains a prime target for the study and application of antiepileptogenic therapy. A large number of clinical trials have aimed at the prevention of PET. All failed to either prevent or alter the period of epileptogenesis associated with TBI.7,8,10
1 : A population-based study of seizures after traumatic brain injuries. N Engl J Med 1998; 338:20–24Crossref, Medline, Google Scholar
2 : Long-term risk of epilepsy after traumatic brain injury in children and young adults: a population-based cohort study. Lancet 2009; 373:1105–1110Crossref, Medline, Google Scholar
3 : Risk of epilepsy after traumatic brain injury: a retrospective population-based cohort study. J Neurol Neurosurg Psychiatry 2013; 84:441–445Crossref, Medline, Google Scholar
4 : A population-based study of risk of epilepsy after hospitalization for traumatic brain injury. Epilepsia 2010; 51:891–898Crossref, Medline, Google Scholar
5 : Clinical epidemiology of post-traumatic epilepsy in a group of Chinese patients. Seizure 2012; 21:322–326Crossref, Medline, Google Scholar
6 : Epileptic seizures and epilepsy: definitions proposed by the International League Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE). Epilepsia 2005; 46:470–472Crossref, Medline, Google Scholar
7 : Post-traumatic epilepsy: an overview. Clin Neurol Neurosurg 2006; 108:433–439Crossref, Medline, Google Scholar
8 : Post-traumatic epilepsy. Handbook Clin Neurol 2012; 108:585–599Crossref, Medline, Google Scholar
9 : Head trauma and epilepsy, in Jasper's Basic Mechanisms of the Epilepsies. Edited by Noebels JLAvoli MRogawski MA. Bethesda, MD, National Center for Biotechnology Information (US), 2012. [Internet]Crossref, Google Scholar
10 Cesnik E, Casetta L, Granieri E. Post-traumatic epilepsy: review. J Neurol Neurophysiol 2013; S2:009.Google Scholar
11 : The risks of epilepsy after traumatic brain injury. Seizure 2000; 9:453–457Crossref, Medline, Google Scholar
12 : Concussive convulsions: emergency department assessment and management of a frequently misunderstood entity. Acad Emerg Med 2001; 8:296–298Crossref, Medline, Google Scholar
13 : Risk of seizure recurrence after the first late post-traumatic seizure. Arch Phys Med Rehabil 1997; 78:835–840Crossref, Medline, Google Scholar
14 : Factors predicting prognosis of epilepsy after presentation with seizures. Ann Neurol 2000; 48:833–841Crossref, Medline, Google Scholar
15 : Clinical semiology of frontal lobe seizures, in Frontal lobe seizures and epilepsies. Edited by Chauvel PDelgado-Escueta AVHalgren E. New York, Raven Press Ltd, 1992, pp 3–58Google Scholar
16 : Neurophysiologic and neuroradiologic features of intractable epilepsy after traumatic brain injury in adults. Arch Neurol 2000; 57:1611–1616Crossref, Medline, Google Scholar
17 : Analyzing risk factors for late post-traumatic seizures: a prospective, multicenter investigation. Arch Phys Med Rehabil 2003; 84:365–373Crossref, Medline, Google Scholar
18 : Post-traumatic epilepsy: neuroradiologic and neuropsychological assessment of long-term outcome. Epilepsia 2003; 44:569–574Crossref, Medline, Google Scholar
19 : Risk factors for late-onset seizures related to cerebral contusions in adults with a moderate traumatic brain injury. Clin Neurol Neurosurg 2011; 113:469–471Crossref, Medline, Google Scholar
20 : Intractable epilepsy and craniocerebral trauma: analysis of 163 patients with blunt and penetrating head injuries sustained in war. Injury 2012; 43:2132–2135Crossref, Medline, Google Scholar
21 : Evaluation of seizure-like episodes in survivors of moderate and severe traumatic brain injury. J Head Trauma Rehabil 2004; 19:290–295Crossref, Medline, Google Scholar
22 : Seizure localization and pathology following head injury in patients with uncontrolled epilepsy. Neurology 1995; 45:2051–2057Crossref, Medline, Google Scholar
23 : There is differential loss of pyramidal cells from the human hippocampus with survival after blunt head injury. J Neuropathol Exp Neurol 2003; 62:272–279Crossref, Medline, Google Scholar
24 : Quantitative structural changes in white and gray matter 1 year following traumatic brain injury in rats. Acta Neuropathol 2002; 103:607–614Crossref, Medline, Google Scholar
25 : Neuronal and glial cell number in the hippocampus after experimental traumatic brain injury: analysis by stereological estimation. J Neurotrauma 2003; 20:929–941Crossref, Medline, Google Scholar
26 : Hippocampal cell loss in post-traumatic human epilepsy. Epilepsia 2006; 47:1373–1382Crossref, Medline, Google Scholar
27 : Epilepsy after head injury: an overview. Epilepsia 2009; 50(Suppl 2):4–9Crossref, Medline, Google Scholar
28 : Traumatic brain injury: risks of epilepsy and implications for medicolegal assessment. Epilepsia 2012; 53(Suppl 4):43–47Crossref, Medline, Google Scholar
29 : Epilepsy after penetrating head injury. I. Clinical correlates: a report of the Vietnam Head Injury Study. Neurology 1985; 35:1406–1414Crossref, Medline, Google Scholar
30 : Penetrating injuries in the Vietnam war. Traumatic unconsciousness, epilepsy, and psychosocial outcome. Neurosurg Clin N Am 1995; 6:715–726Crossref, Medline, Google Scholar
31 : Correlates of post-traumatic epilepsy 35 years following combat brain injury. Neurology 2010; 75:224–229Crossref, Medline, Google Scholar
32 : Adenosine A1 receptor gene variants associated with post-traumatic seizures after severe TBI. Epilepsy Res 2010; 90:259–272Crossref, Medline, Google Scholar
33 : MTHFR C677T genotype as a risk factor for epilepsy including post-traumatic epilepsy in a representative military cohort. J Neurotrauma 2011; 28:1739–1745Crossref, Medline, Google Scholar
34 : Genetic variability in glutamic acid decarboxylase genes: associations with post-traumatic seizures after severe TBI. Epilepsy Res 2013; 103:180–194Crossref, Medline, Google Scholar
35 : Unraveling the genetics of common epilepsies: approaches, platforms, and caveats. Epilepsy Behav 2013; 26:229–233Crossref, Medline, Google Scholar
36 : Post-traumatic epilepsy: hemorrhage, free radicals and the molecular regulation of glutamate. Neurochem Res 2009; 34:688–697Crossref, Medline, Google Scholar
37 : Imaging of head trauma, in Imaging of the nervous system. Edited by Latchaw REKucharczyk JMoseley ME. Philadelphia, PA, Elsevier Mosby, Inc, 2005, pp 869–904Google Scholar
38 : Advances in understanding the process of epileptogenesis based on patient material: what can the patient tell us? Epilepsia 2003; 44(Suppl 12):60–71Crossref, Medline, Google Scholar
39 : The role of inflammation in epileptogenesis. Neuropharmacology 2013; 69:16–24Crossref, Medline, Google Scholar
40 : Immune mechanisms in epileptogenesis. Front Cell Neurosci 2013; 7:195Crossref, Medline, Google Scholar
41 McNamara JO, Huang YZ, Leonard AS. Molecular signaling mechanisms underlying epileptogenesis. Sci STKE 2006; 2006:re12.Google Scholar
42 : Brain inflammation in epilepsy: experimental and clinical evidence. Epilepsia 2005; 46:1724–1743Crossref, Medline, Google Scholar
43 : Epileptogenesis in the dentate gyrus: a critical perspective. Prog Brain Res 2007; 163:755–773Crossref, Medline, Google Scholar
44 : The neurobiology of epilepsy. Curr Neurol Neurosci Rep 2007; 7:348–354Crossref, Medline, Google Scholar
45 : Animal models of head trauma. NeuroRx 2005; 2:410–422Crossref, Medline, Google Scholar
46 : Experimental models of traumatic brain injury: do we really need to build a better mousetrap? Neuroscience 2005; 136:971–989Crossref, Medline, Google Scholar
47 : From traumatic brain injury to post-traumatic epilepsy: what animal models tell us about the process and treatment options. Epilepsia 2009; 50(Suppl 2):21–29Crossref, Medline, Google Scholar
48 : The role of advanced MR imaging findings as biomarkers of traumatic brain injury. J Head Trauma Rehabil 2010; 25:267–282Crossref, Medline, Google Scholar
49 : Prospects for imaging-related biomarkers of human epileptogenesis: a critical review. Biomarkers Med 2011; 5:599–606Crossref, Medline, Google Scholar
50 : Multimodal neuroimaging: potential biomarkers for response to antiepileptic drugs? Epilepsia 2013; 54(Suppl 2):67–70Crossref, Medline, Google Scholar
51 : Diffusion tensor imaging in late post-traumatic epilepsy. Epilepsia 2005; 46:1465–1471Crossref, Medline, Google Scholar
52 : Molecular and diffusion tensor imaging of epileptic networks. Epilepsia 2008; 49(Suppl 3):15–22Crossref, Medline, Google Scholar
53 : Diffusion-based magnetic resonance imaging and tractography in epilepsy. Epilepsia 2008; 49:189–200Crossref, Medline, Google Scholar
54 : Group-specific regional white matter abnormality revealed in diffusion tensor imaging of medial temporal lobe epilepsy without hippocampal sclerosis. Epilepsia 2010; 51:529–535Crossref, Medline, Google Scholar
55 : Evaluation of cerebral white-matter micro-structural alterations in patients with medically refractory epilepsy using diffusion tensor tractography. Epilepsy Res 2013; 107:82–90Crossref, Medline, Google Scholar
56 : Post-traumatic epilepsy: What's contusion got to do with it? Epilepsy Curr 2012; 12:87–91Crossref, Medline, Google Scholar
57 : Predicting post-traumatic epilepsy with MRI: prospective longitudinal morphologic study in adults. Epilepsia 2005; 46:1472–1481Crossref, Medline, Google Scholar
58 : Blood-brain barrier breakdown as a therapeutic target in traumatic brain injury. Nat Rev Neurol 2010; 6:393–403Crossref, Medline, Google Scholar
59 : Blood-brain barrier dysfunction and epilepsy: pathophysiologic role and therapeutic approaches. Epilepsia 2012; 53:1877–1886Crossref, Medline, Google Scholar
60 Tomkins O, Feintuch A, Benifla M et al. Blood-brain barrier breakdown following traumatic brain injury: a possible role in post-traumatic epilepsy. Cardiovasc Psychiatry Neurol 2011; 2011:765923.Google Scholar

