
Buprenorphine Does Not Aggrevate Ischemic Neuronal Injury in Experimental Focal Cerebral Ischemia
However, available data suggest increased risk of cerebrovascular events in opioid-dependent patients. 3 – 6 In addition, focal neurological deficits have been reported to occur immediately following heroin injection and up to and beyond 24 hours after heroin abuse. Thus, an agent offering safety in regard to neurological function would be quite valuable for opioid-dependent patients. It has recently been shown that buprenorphine can cause some alterations on the opioid receptors in rat brain regions, 7 which are important indicators of infarct extension in ischemic neuronal damage. 8 Given these in vitro findings indicating the neurotoxic role of buprenorphine, we aim to test the hypothesis of whether buprenorphine is neurotoxic in vivo during cerebral ischemia.
METHOD
All experimental procedures were carried out with governmental approval according to local guidelines for the care and use of laboratory animals. Adult male Wistar rats weighing 330 g to 370 g were assigned to the following experiments and groups: intraperitoneal administration of a) 2 mg/kg buprenorphine hydrochloride (Sigma) dissolved in saline (N=11 animals), b) 0.2 ml vehicle (0.09percnt; NaCl) (N=9 animals), starting at the onset of transient focal cerebral ischemia.
Induction of Ischemia
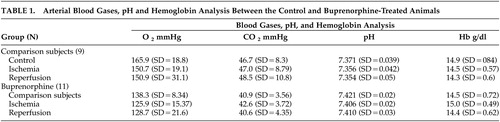
Animals were anesthesized with 4 vol% isoflurane (30% O 2 , remainder N 2 O). Rectal temperature was maintained between 36.5 and 37.0°C using a feedback-controlled heating system. During the experiments, cerebral blood flow was measured by Laser Doppler Flowmetry (LDF) using a flexible fiber optic probe (Perimed, Stockholm), which was attached to the intact skull overlying the middle cerebral artery territory (1 mm posterior/5 mm lateral from bregma). Laser doppler flowmetry changes were monitored up to 90 minutes after the onset of ischemia. Focal cerebral ischemia was induced using an intraluminal filament technique. 9 A midline neck incision was made, and the common carotid artery (CCA) was temporarily closed with two microvascular clips (FE691, Aesculap, Tuttlingen, Germany). A 4–0 nylon monofilament (Ethilon; Ethicon, Norderstedt, Germany) coated with silicon resin (Xantopren, Bayer Dental, Osaka, Japan) was introduced through a small incision into the common carotid artery and advanced distal to the carotid bifurcation for transient occlusion of the middle cerebral artery. The filament was removed after 90 minutes,and the CCA incision was closed by single interrupted sutures,using a 10–0 silk suture. We discontinued anesthesia and placed animals into their home cages. Arterial blood gases, pH, hemoglobin, mean arterial blood pressure, and heart rate were measured during ischemia and reperfusion.
Infarct Volume Measurements: Toluidine Blue Staining
The animals were deeply anesthetized with isoflurane 24 hours after middle cerebral artery occlusion, and then decapitated. Brains were immersion-fixed in 3.8% buffered formaldehyde for 5 days and automatically processed for embedding in paraffin (Bavimed 2050Z2; Haska AG, Bern, Switzerland) through a series of ethanol baths (70–100%) into xylol and paraffin for over 21 hours. Serial coronal sections (10 μm thick, three brains per block) were cut at 1-mm intervals through the forebrain and stained with toluidine blue ( Figure 1a ). The boundaries of the infarcted cerebral cortex and neostriatum were marked by an unbiased observer and traced from a monitor (at 21x magnification) onto a digitizing tablet interfaced to an image analysis system (DiaSys Datalab; H. Meyer, Thöringen, Switzerland). The infarct volumes were calculated by numeric integration of the infarct areas on sequential slices without correction for edema.
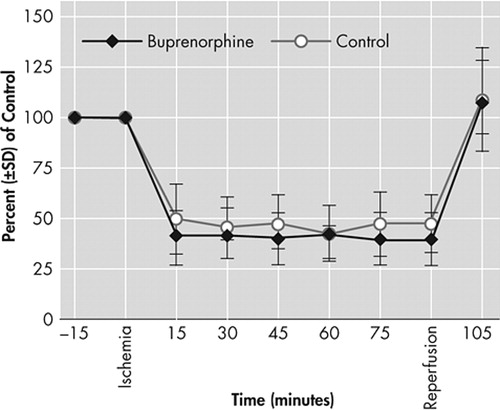
Measurements taken during 90 minutes of intraluminal thread occlusion and immediately after reperfusion in animals treated with vehicle solution or buprenorphine (2 mg/kg). No differences in laser doppler flowmetry values are detected between animal groups. Values are means (SD)
Statistics
All values are given as means (SD). We compared differences between groups by using independent sample t tests. We considered p values <0.05 to indicate statistical significance.
RESULTS
Laser Doppler Flow and Physiological Parameters
Laser Doppler flow values decreased to approximately 40% of preischemic levels immediately after thread insertion. After thread retraction, laser Doppler flowmetry values slightly increased above preischemic control levels. Again, no differences were detected between different animal groups ( Figure 2 ). Arterial blood gas parameters and pH were kept within the physiological range ( Table 1 ). The mean arterial blood pressure and the heart rate moved in a physiological range prior to the occlusion and 90 minutes after reperfusion, and there were no significant differences between animal groups.
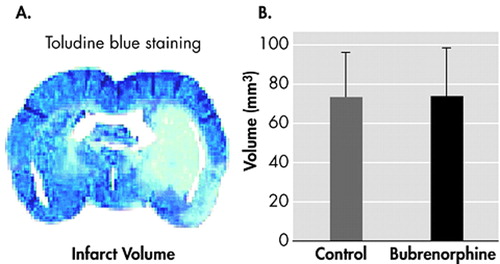
Infarcts were obtained in the 2 mg/kg, or vehicle-treated groups. Infarct volumes determined by Toluidine blue staining (A). Infarct volumes of mice subjected to transient cerebral ischemia. Note that 2 mg/kg buprenorphine does not lead to a increasement in infarct size (B). Data are given as means (SD)
 |
Infarct Size
We obtained reproducible brain infarcts in the 2 mg/kg of buprenorphine or 0.2 ml of vehicle treated groups ( Figure 1 ). Intraperitoneal administration of 2 mg/kg buprenorphine had no effect on the infarct volume compared with control animals ( Figure 1 ).
DISCUSSION
Buprenorphine has been in clinical use as an analgesic agent for several decades. It is increasingly used in maintenance therapy of opioid dependence. Opioid-dependent patients are at higher risk of cerebrovascular events and focal neurological deficits. 3 – 6 In vitro studies available to date strongly suggest neurotoxic effects of buprenorphine. 10 , 11 However, this in vivo study does not indicate neurotoxic effects of buprenorphine in focal cerebral ischemia model.
A broad spectrum of neuropathological changes has been encountered in the brains of heroin abusers. 3 These changes have been associated either with infections (either due to bacterial spread from bacterial endocarditis, mycoses, or from human immunodeficiency virus infection) or to the other complications resulting from hypoxic/ischemic injuries and related neuronal damage. However, thromboembolism, vasculitis, septic emboli, hypotension, and positional vascular compression are some of the mechanisms hypothesized for respiratory depression and stroke in heroin abusers. 3 Given that the infarct sizes between the buprenorphine and saline-treated groups were not different from each other, the present study demonstrates that single administration of buprenorphine does not cause neurotoxicity in mice with a focal permanent cerebral ischemia model.
It is difficult to explain the reasons behind the difference in findings of this in vivo study and the previous ones in vitro. Yet, while the opioidergic system in the brain has been implicated in the pathophysiology of cerebral ischemia, 8 , 12 the studies indicating the neuroprotective role of opioid receptor (KOR) agonists/antagonists are still controversial. Moreover, although recent studies indicated the role of kappa receptor agonists as neuroprotectants in cerebral ischemia, 12 – 14 other studies suggested the role of kappa receptors as mediators for ischemia aggravating effect for endogenous opioids. 15 – 18 Unfortunately, human studies evaluating the neuroprotective role of opioid receptor antagonists failed to find any treatment benefit in clinical studies. 19 – 21
Thus, in regard to these controversial results of receptor targeted treatments, it is difficult to explain the difference of this in vivo study from previous in vitro studies only with its kappa receptor antagonist activity which may have balanced its neuroprotection. Moreover, if the same finding can be replicated by the chronic administration of buprenorphine in vivo, adaptative changes leading to receptor up- or down-regulation in opioid receptors may also be discussed as a contributing factor preventing the development of ischemic neuronal damage.
In conclusion, this study provides strong preclinical evidence for the safety of using buprenorphine during the postoperative period following ischemic events, as well as for an alternative maintenance therapy of opioid-dependent patients wherein the risk of cerebrovascular events is increased.
1 . Cowan A, Lewis JW, MacFarlane IR: Agonist and antagonist properties of buprenorphine, a new antinociceptive agent. Br J Pharmacol 1977; 60:537–545Google Scholar
2 . Heel RC, Brodgen RN, Speight TM, et al: Buprenorphine: a review of its pharmacological properties and therapeutic efficacy. Drugs 1979; 17:81–110Google Scholar
3 . Neiman J, Haapaniemi HM, Hillbom M: Neurological complications of drug abuse: pathophysiological mechanisms. Eur J Neurol 2000; 7:595–606Google Scholar
4 . Bartolomei F, Nicoli F, Swiader L, et al: Ischemic cerebral vascular stroke after heroin sniffing: a new case. Presse Med 1992; 6:983–986Google Scholar
5 . Jensen R, Olsen TS, Winther BB: Severe non-occlusive ischemic stroke in young heroin addicts. Acta Neurol Scand 1990; 81:354–357Google Scholar
6 . Brust JC, Richter RW: Stroke associated with addiction to heroin. J Neurol Neurosurg Psychiatry 1976; 39:194–199Google Scholar
7 . Belcheva MM, Ho MT, Ignatova EG, et al: Buprenorphine differentially alters opioid receptor adaptation in rat brain regions. J Pharmacol Exp Ther 1996; 277:1322–1327Google Scholar
8 . Boutin H, Dauphin F, MacKenzie ET, et al: Differential time-course decreases in nonselective, mu-, delta-, and kappa-opioid receptors after focal cerebral ischemia in mice. Stroke 1999; 30:1271–1277Google Scholar
9 . Koizumi J, Yoshida Y, Nakazawa T, et al: Experimental studies of ischemic brain edema, I: a new experimental model of cerebral embolism in rats in which recirculation can be introduced in the ischemic area. Jpn J Stroke 1986; 8:1–8Google Scholar
10 . Kugawa F, Ueno A, Aoki M: Apoptosis of NG108–15 cells induced by buprenorphine hydrochloride occurs via the caspase-3 pathway. Biol Pharm Bull 2000; 23:930–935Google Scholar
11 . Kugawa F, Arae K, Ueno A, et al: Buprenorphine hydrochloride induces apoptosis in NG108–15 nerve cells. Eur J Pharmacol 1998; 347:105–112Google Scholar
12 . Zabramski JM, Spetzler RF, Selman WR, et al: Naloxone therapy during focal cerebral ischemia evaluation in a primate model. Stroke 1984; 15:621–627Google Scholar
13 . Kusomoto K, Mackay KB, McCulloch J: The effect of the kappa-opioid receptor agonist CI-977 in a rat model of focal cerebral ischemia. Brain Res 1992; 576:147–151Google Scholar
14 . Baskin DS, Widmayer MA, Browning JL, et al: Evaluation of delayed treatment of focal cerebral ischemia with three selective kappa-opioid agonists in cats. Stroke 1994; 25:2047–2054Google Scholar
15 . Krumins SA, Faden AI: Traumatic injury alters opiate receptor binding in spinal cord. Ann Neurol 1986; 19:498–501Google Scholar
16 . Herman BH, Goldstein A: Antinociception and paralysis induced by intrathecal dynorphin A. J Pharmacol Exp Ther 1985; 232:27–32Google Scholar
17 . Faden AI, Molineaux CJ, Rosenberger JG, et al: Increased dynorphin immunoreactivity in spinal cord after traumatic injury. Regul Pept 1985; 11:35–41Google Scholar
18 . Scavini S, Rozza A, Bo P, et al: Opioid receptor changes and neurophysiological alterations during cerebral ischemia in rabbits. Stroke 1990; 21:943–947Google Scholar
19 . Fallis RJ, Fisher M, Lobo RA: A double blind trial of naloxone in the treatment of acute stroke. Stroke 1984; 15:627–629Google Scholar
20 . Perraro F, Tosocini G, Pertoldi F, et al: Double-blind placebo controlled trial of naloxone on motor deficits in acute cerebrovascular disease. Lancet 1984; 1:915Google Scholar
21 . Clark WM, Raps EC, Tong TC, et al: Cervene (nalmefene) in acute ischemic stroke: final results of a phase III efficacy study. Stroke 2000; 31:1234–1239Google Scholar

