
The EEG and Cocaine Sensitization
Abstract
The author presents the hypothesis that reduced delta EEG power observed in cocaine withdrawal is related to changes in dopamine (DA) transmission related to cocaine sensitization. Evidence for this hypothesis includes the topographic anatomical correspondence between the putative site of delta generation and the cortical terminal field of the mesotelencephalic DA system, as well as the laminar distribution and ultrastructural features of DA terminals in frontal cortex that appear to be adapted to the modulation of the delta rhythm, a global forebrain EEG mode. The effect of DA on membrane conductances of individual pyramidal neurons also suggests that DA exerts a significant influence on delta power by modulating the transition between global and local EEG modes. Access to a neural correlate of sensitization via noninvasive EEG methodology could be useful in investigating the relationship of stimulant sensitization to the clinical syndrome of cocaine dependence.
The physiologic relevance of the electroencephalogram (EEG) as a reflection of neural activity1 and the evidence of abnormal neural activity in cocaine dependence2–4 suggest the possibility that the EEG might reflect abnormal neurobiology in cocaine dependence. The hypothesis presented in this article links abnormal EEG findings observed in cocaine dependence2,5–10 to stimulant sensitization. Although such a discussion is necessarily speculative, it might ultimately prove to be useful given the potential advantages of the EEG as a means of studying cocaine dependence in humans. The EEG is noninvasive and inexpensive, and quantitative analysis of the EEG (qEEG) is based on digital technology that is constantly growing in analytic power and accessibility. Clinically, the EEG is sensitive to psychiatric conditions such as depression or attention-deficit/hyperactivity disorder (ADHD) that are often comorbid with cocaine dependence.11,12 Pretreatment qEEG has shown a correlation with subsequent psychotropic drug response in patients with a variety of psychiatric disorders evaluated prospectively.11 Using the EEG to gain access to a neural correlate of sensitization could possibly be useful in the investigation of its relationship to the clinical syndrome of cocaine dependence, and ultimately in the development or selection of medications according to a hypothesis of stimulant sensitization.
Stimulant sensitization is observed behaviorally in animal models as a progressive enhancement of the locomotor response to cocaine or amphetamine with repeated exposure.13–15 The induction of behavioral sensitization to amphetamine has also been recently demonstrated in humans.16,17 An extensive literature on work in animals suggests that behavioral sensitization to cocaine or amphetamine corresponds to persistent, characteristic neuroadaptions in brain structures that subserve reward-mediated behavior, including the mesotelencephalic dopamine (DA) system.13–15
The more consistent neurochemical findings suggestive of persistent neuroadaptive changes in stimulant sensitization include increased DA efflux in the nucleus accumbens (NAc) in response to a cocaine or amphetamine challenge14,15,18,19 and enhancement of signal transduction through DA D1 receptors15,20–23 and inhibition of NAc neurons.24–26 The latter two phenomena are of particular interest with regard to sensitization and the EEG because they can manifest on a tonic, continual basis, and therefore they occur on a temporal scale consistent with the spontaneous resting EEG. The robust consistency of the phenomenon of stimulant sensitization in animal experimental paradigms and the apparent involvement of neural systems implicated in reward and motivation have led to significant interest in the possible role of sensitization in the clinical syndrome of cocaine dependence.13,14,27,28 It has been suggested that a sign of sensitization, namely increased DA efflux in the NAc, is a correlate of salience and motivational relevance, and that its augmentation over time in response to cocaine or cocaine-related cues with sensitization reflects the increasing attentional and motivational focus on cocaine and cocaine-related stimuli.28–30
In the following discussion, the literature on the neurobiology of cocaine sensitization is reviewed in the context of the generation and dopaminergic modulation of the EEG, and the hypothesis is presented that the EEG power spectral profile observed in cocaine withdrawal reflects changes in DA neurotransmission that are associated with stimulant sensitization.
COCAINE AND THE EEG
The reported acute and chronic qEEG effects of cocaine in animals and humans generally involve some combination of enhanced fast activity, diminished slow activity, and broad reductions in EEG amplitudes across the entire power frequency spectrum. These are EEG states to which terms such as desynchronization or arousal31 have been applied. That common attributes have been observed between EEG states following acute and chronic exposure is consistent with the conceptualization of sensitization as a “proponent” process.32 In a proponent process, similarities exist between the state following acute drug administration and the state of withdrawal following chronic drug administration. In the case of cocaine, specific phenomena with observed consistency between the acute and chronic states include enhanced transmission through DA D1 receptors and inhibition of NAc neurons.15
The effects of acute and chronic exposure may be viewed as generally consistent with the transition from global to local EEG modes.33Global EEG modes tend to originate from sources with a relatively widely dispersed topographic distribution and have high amplitudes and low frequencies. Activity in the “slow” bandwidths delta and theta occurring in normal awake humans and the eyes-closed occipital alpha rhythm are examples of global EEG modes. Functionally, global EEG modes are thought to serve the purpose of gating input or motor output and coordinating activity across widely distributed cortical regions. The apparent gating of sensory input in the visual cortex by the occipital alpha rhythm is an example of the gating function attributed to a global EEG mode. Global modes might also serve the purpose of facilitating coordinated activity between relatively widely distributed cortical expanses, such as the putative role of the theta rhythm in enhancing transmission between temporal allocortex and frontal neocortex.34,35Local EEG modes are higher in frequency, lower in amplitude, and distributed over a more limited topographic area. Functionally, local EEG modes tend to correspond to tasks that involve attending to incoming stimuli or outputting motor action. An example of a local EEG mode would be the appearance of beta frequency activity over visual cortex during a target detection task, or over contralateral motor cortex during a manual movement task.31
Acute administration of cocaine in rats reportedly results in diminished slow EEG power and overall amplitudes and increased beta frequency power.36–39 These EEG effects appear to be mediated by DA to an important extent, as evidenced by their temporal correspondence to DA levels in the prefrontal cortex and their diminution by D1 antagonist.36,37 Several findings are consistent with a role of increased transduction through D1 receptors in sensitization. Reduced global EEG amplitudes have been observed with the administration of either cocaine (10 mg/kg) or a selective D1 agonist38,39 and in the spontaneous EEG in drug-free rats previously exposed to cocaine according to a schedule designed to produce sensitization.40 Cocaine-sensitized rats also reportedly show a greater relative decrement of EEG power in the delta and theta bands in response to an acutely administered dose of cocaine.40 The possibility that the greater reduction in slow activity in response to acute cocaine in sensitized animals might reflect increased DA signal transduction receives support from an animal study on rats involving ventral tegmental area (VTA) stimulation. Leung and Yim41 reported on a delta frequency rhythm recorded directly from the NAc, which they termed accumbens delta, that was desynchronized by electrical stimulation of the VTA; this effect was blocked by haloperidol.
In humans, acute administration of cocaine is most commonly reported to enhance beta frequency activity.42–44 As has been reported for animals,40 the acute effects of cocaine on slow EEG activity in humans may differ as a function of prior exposure to cocaine. Relatively inexperienced intranasal cocaine users challenged with intravenous (IV) cocaine have reportedly evidenced a transient delta increase in addition to the expected, and more persistent, beta increase in the first 5 minutes following the injection,42 which coincides with the expected time of peak euphoria.45 More experienced IV users tested in a later study did not evidence a delta increase.44 The transient enhancement of delta activity in relatively naive users, but not in chronic users, could represent a neuroadaptation due to chronic use, possibly sensitization, resulting in a progressive diminution of activity in global EEG modes.
Five studies, summarized in Table 1, have reported on the qEEG in drug-free subjects with prior histories of chronic cocaine use compared with normal control subjects.5–9 In the four of these studies in which reduced delta and theta power were noted relative to values derived from normative databases,5–7,9 visual inspection and automatic detection with exclusion of artifactual epochs were used to control for EEG artifact. The single study that did not note a slow wave deficit8 relied on a mathematical algorithm to reduce the effect of ocular artifact without the actual exclusion of any artifactual epochs, a methodologic factor that might have been particularly likely to have affected delta frequency findings; also, the lack of a normative database and inclusion of eyes-open data might have limited sensitivity. In four of the studies listed in Table 1, a relative or absolute beta increase was seen.5,6,8,9 Discrepant findings regarding beta activity may have been due to electrophysiological heterogeneity between subgroups of subjects7 and differences related to route of administration. Use of cocaine by the IV route is reportedly associated with greater beta power than cocaine smoking.46 The study that reported no overall beta excess in crack cocaine users rigorously excluded subjects with a history of any IV drug use7 and found beta excess only in an electrophysiologic subgroup, defined by cluster analysis of EEG variables, that was characterized clinically by early dropout from treatment.10 Alpha relative power is reportedly increased in the studies that found an absolute deficit of slow activity,5–7 and the finding apparently relates more to the slow wave deficit than to an absolute alpha excess. Only one study,5 which was a pilot sample of 7 subjects, found an absolute alpha power elevation; this finding might have been attributable to the relatively high depressive comorbidity in that sample in view of reports of increased alpha power in depression.8,11
These results in human subjects or animals chronically or acutely exposed to cocaine appear to share the attribute of bias toward the transition from global to local EEG modes, which might be manifested as a diminution of slow or an enhancement of fast EEG activity. Overall, there appears to be significant agreement among the published studies of drug-abstinent, cocaine-dependent humans in finding a slow wave deficit, along with a beta excess in at least a subgroup of subjects. The apparent similarity of the acute and chronic states is consistent with the conceptualization of cocaine sensitization as a proponent process.32 In the discussion that follows, the hypothesis is presented that cocaine sensitization results in a transition from global to local EEG modes in the frontal cortical terminal fields of the mesotelencephalic DA projection, and that this transition is reflected by diminished delta, a global forebrain EEG mode.
The observation of an association of beta EEG power, a local EEG mode, with treatment failure10 could logically be related to a sensitization hypothesis. Increased frontal beta activity has also been reported to correlate prospectively with treatment failure in two independent samples of subjects with alcohol dependence.47,48 In view of the observation that VTA stimulation in animals enhances beta power and diminishes delta power,41,49 the relative enhancement of beta EEG power in treatment dropouts is possibly consistent with hypotheses linking substance dependence and neural sensitization in the mesotelencephalic DA system.14,15,18
The discussion that follows, however, will focus on the finding of decreased delta EEG power as a possible correlate of cocaine sensitization. In our studies on crack cocaine dependence,2,5,7,10 as well as that of Roemer et al.,6 the delta deficit has been the most statistically abnormal finding and appears to be generally shared across subgroups of patients. A delta deficit has not generally been reported in conditions that could potentially confound EEG results in cocaine dependence, such as alcohol or cannabis dependence or human immunodeficiency virus infection.11,50–52 A beta excess, on the other hand, is a frequently reported feature in alcohol dependence.8,11 The abnormal delta findings in cocaine dependence persist for at least 6 months,2 which is a time frame consistent with the persistence of neural sensitization.14 We have not been able to determine the persistence of the beta excess that we have seen in the subgroup of subjects who prematurely depart from treatment, since these subjects were not available for retesting after an extended drug-free interval. The discussion that follows will therefore focus on the possible significance of the observation of reduced delta EEG power in cocaine withdrawal as a possible correlate of cocaine sensitization.
FUNCTIONAL SIGNIFICANCE OF THE AWAKE DELTA EEG RHYTHM
The human brain generates delta frequency EEG activity in a variety of neurological and behavioral states, and it is important to specify that the delta EEG power of interest in this discussion is that observed in awake, neurologically normal adults. Delta power can comprise more than 20% of total EEG power in frontal leads in young adults in multiply replicated normative studies.53–58 The conventional EEG literature emphasizes pathologically excessive delta as a consequence of cortical deafferentation or subcortical inflammatory processes34 but provides little discussion of the significance of a delta deficit. The term delta is used herein to refer to the delta EEG rhythm that is observed in neurologically normal, awake adults; it is not the delta seen in sleep, in infancy, or in the presence of neurologic pathology. There is substantial evidence that delta can appear as a correlate of complex cognitive processes. Delta EEG power has been observed to be increased in normal subjects performing calculations,59,60 reaction time tasks,61 abstract thought,62 P300 paradigms,63,64 and a delayed match-from-sample paradigm.65
Delta EEG activity is an example of a global neocortical processing mode. Global EEG processing modes involve low-frequency EEG activity spanning relatively large cortical regions, as opposed to local modes involving high-frequency EEG activity distributed over smaller cortical areas.33 Global modes have been hypothesized to serve the purpose of integration across diverse cortical sites by synchronizing coherent activity and phase coupling across widely spatially distributed neural assemblies. The observation of increased delta EEG power in tasks spanning processes such as working memory or abstraction59–65 is consistent with a role of delta in the process of integration of activity across association cortex. Global modes might also serve the function of gating extraneous stimuli from gaining access to global assemblies of coherent EEG activity.1,66 A gating function has been suggested for delta power in the functional disconnection of the cortex from extraneous thalamic inputs, analogous to the role postulated for alpha in quiescent states of visual sensory cortex.67 This functional cortical deafferentation has been hypothesized to serve the process of selective attention to information represented in association cortex during states of “internal concentration.”59,60 Delta in normal awake humans can apparently be a correlate of the process of allocating attention and limiting access of extraneous stimuli during states in which the cortex is processing its own output. Therefore, a delta deficit could conceivably relate to vulnerability toward the preemption of attention by craving and drug-related cues in cocaine dependence.
With regard to the functional role of delta power, it is interesting to note that the other group in our database that most consistently appears to have a delta deficit is subjects with ADHD.12 The apparently shared feature of a delta deficit in cocaine dependence and ADHD coexists with other evidence of a relationship between the two disorders, which includes a high comorbid incidence, a familial association between cocaine dependence and ADHD,68 and the common general behavioral features of impulsivity and disturbed regulation of attention.
There is apparently a significant commonality between normal awake delta EEG activity and the P300. The P300 is an evoked response to stimuli that are unexpected, infrequent, or motivationally relevant. In a typical P300 paradigm, a subject is asked to respond to a relatively infrequent target stimulus, as opposed to a more common nontarget stimulus. The amplitude of the P300 increases with the motivational relevance of the task or the salience of the target stimulus, and P300 amplitude reflects the allocation of attentional resources. The psychological processes spanned by P300 paradigms include working memory, motivation, attention, and the appreciation of context.69 P300 amplitude has been observed to correlate positively with delta EEG power.64,70,71 Delta EEG activity is selectively enhanced in epochs immediately following target stimuli presented in the context of a P300 paradigm, but it is not enhanced when the same stimuli are presented in a context that does not elicit a P300 response.63,64 The increased cortical resonance or frequency-specific enhancement of EEG in the delta range during cognitive and attentional processes suggests that delta is a correlate of these processes.
GENERATION AND DOPAMINERGIC MODULATION OF THE AWAKE DELTA EEG RHYTHM
Neuroimaging Studies
Neuroimaging studies in humans provide evidence supporting a functional relationship of the mesotelencephalic DA projection to the delta rhythm. One line of evidence linking DA and delta is the shared medial frontal cortical location of the terminal fields of the mesotelencephalic DA system and the apparent site of delta generation indicated by EEG source localization and metabolic imaging. Studies using dipole modeling place the site of delta generation in anterior medial frontal cortex.62,72
Likewise, studies correlating positron emission tomography (PET) and EEG indicate a positive correlation between PET metabolism and waking delta activity in medial frontal cortex in both schizophrenic73 and normal subjects.74 The positive correlation of PET metabolism and waking delta in medial frontal cortex contrasts with the negative relationship of the delta of slow wave sleep to PET metabolism75 or regional cerebral blood flow in the medial frontal cortex.76 These apparently opposite relationships of delta to metabolism and blood flow in medial frontal cortex in sleep versus awake states emphasize the functional distinction between the normal awake delta rhythm and delta of slow wave sleep.
In view of the observed diminution of delta power in cocaine dependence, and the apparently positive correlation of PET metabolism to normal awake delta power in medial frontal cortex, reduced frontal cortical metabolic activity would be expected to correlate with the reduced delta EEG power reported in cocaine withdrawal. Consistent with such a formulation are reports that cocaine-dependent subjects evaluated after up to 3 months of drug abstinence had decreased metabolism measured by PET, localized mainly to the frontal lobes,3,77 and that perfusion deficits in frontal regions were evident on single-proton emission computed tomography after up to 6 months of cocaine withdrawal.78,79
Figure 1 illustrates a comparison of 2-Hz EEG power between 35 crack cocaine users at 30 days of drug abstinence and 38 normal control subjects, using the technique of low-resolution electromagnetic topographic analysis (LORETA).80,81 LORETA is an inverse solution that estimates the location of EEG sources in three dimensions from the two-dimensional surface EEG recording. The LORETA image contrasts with the much milder anterior-to-posterior gradient of the delta deficit obtained by conventional two-dimensional analysis of the surface EEG.5–7Figure 1 indicates much larger differences from normal in the frontal than the posterior regions. One possible pathophysiologic interpretation of these results is that the frontal location of the delta deficit reflects neuroanatomical localization of the terminal fields of the mesotelencephalic DA system.
Laminar, Ultrastructural, and Electrophysiologic Features of Cortical DA Terminals
The laminar distribution and ultrastructural features of DA synapses in the cerebral cortex also suggest a role for DA in modulating the transition between global and local EEG modes, and therefore in modulation of the delta rhythm, a global forebrain EEG mode. In the human frontal cortex, DA terminals are distributed mainly in layers I, V, and VI.82–84Figure 2 presents a schematic representation of horizontal integration in the cortex. Layer I is a “horizontal” layer, sparsely populated with principal cells that receive input from deep layers of limbic cortex and superficial layers of association cortex.82 Layers V and VI are “output” layers, having pyramidal cells with axons that project to the NAc, VTA, and thalamus, and apical dendrites that ascend to superficial cortical layers.83
The input to layer I neurons arrives via dendrites that branch extensively, predominantly within layer I. The axons of layer I neurons project mainly to layers II and III. The extensive horizontal distribution of dendritic processes and the limited depth of axonal projections are distinctive features supporting the view of layer I as a horizontal layer, apparently adapted to the integration of diverse limbic and association cortical input.84,85 The presence of large numbers of DA receptors in cortical layer I is a feature that distinguishes the brains of humans and primates from those of rodents,84 and it may reflect the greater requirement for integrating global processing across the relatively greater expanses of association cortex in primates or humans relative to other mammals. The slower EEG frequencies of global modes are associated with relatively greater cortical area, and there is a general correlation of slower mean EEG frequencies to higher phylogenetic sophistication.86 Within layer I, DA levels in humans and primates are maximal in medial prefrontal cortex.84 The relatively high density of DA in cortical layer I in humans is consistent with a role for DA in modulating the transition between global and local modes in frontal cortex.84,87
The “triadic complex” is an ultrastructural feature, characteristic of cortical DA synapses, that is most prominent in layers II and III.84,87,88 The triadic complex consists of a DA terminal and an excitatory cortical projection, synapsing together on a principal cell dendrite. DA receptors situated in this way are well adapted to selectively inhibit multiple sources of excitatory input to layers II and III. These inputs include ascending dendrites from pyramidal cells in deeper cortical layers, descending axons of layer I neurons, nonspecific thalamocortical afferents, and horizontal inputs from adjacent association cortex. The capability of the triadic complex to simultaneously modulate and integrate diverse inputs within the restricted vertical extent of cortical layers II and III implies an important function in the process of horizontal integration. The high levels of DA terminals in layer I, along with the triadic complexes with their associated DA terminals in layers II and III, suggest that DA in superficial cortical layers is well adapted to the function of modulating the transition between global and local EEG modes and therefore to the modulation of the delta rhythm, a neocortical forebrain global EEG mode.
The hypothesized role of DA with regard to observation of reduced delta EEG in cocaine withdrawal does not exclude a role for other neurotransmitters. Norepinephrine (NE), in particular, is a logical candidate. NE shares some effects on pyramidal cell membranes with DA,87 and a recent report found a persistent increase in extracellular NE in the NAc after chronic exposure to cocaine.89 The locus ceruleus (LC) contains most of the cell bodies of the NE projections to the cortex, much as the VTA contains the soma of the DA projections to the cortex. The idea of sensitized LC activity resulting from cocaine exposure receives some support from epidemiologic evidence of a comorbid association of panic disorder and cocaine use.90 There are, however, features of the argument linking DA to the observed delta decrease that appear more specific to DA than to NE. One feature is the evidence of a specific role of the DA D1 receptor in the acquisition and expression of sensitization.15,20,21,23,24 Another feature is the evidence for the shared anatomical location of the site of delta generation implied by PET and EEG dipole analysis, and the location of the terminal fields of the cortical DA projections of VTA. The topographic distribution of NE terminals is generally more diffuse, maximal at somatosensory and motor cortex, and is posterior to the more focal and anterior distribution of DA terminals that is maximal at motor, prefrontal, and limbic cortices.87 The relatively high density of DA terminals in superficial cortical layers is a characteristic that distinguishes human from rodent brains, whereas the laminar distribution of NE is a similar gradient of increasing NE receptor density with depth in both species.84 The distinctive laminar and ultrastructural features of DA terminals have been viewed as supporting a larger role for DA than NE in horizontal integration.84,87
Direct evidence of an important role of DA in modulating between global and local EEG modes is provided by studies of the intrinsic membrane properties of pyramidal neurons, which are the putative source of the generation of forebrain delta EEG activity.34,91 Yang et al.92 studied pyramidal cells of layers V and VI of the rat prefrontal cortex and the effects of DA on ionic conductances that determine membrane excitability.83 The majority of pyramidal cells are capable of pacemaker-like rhythmic bursting in the delta range.91,93–95 The pyramidal cells of cortical layers V and VI have ascending apical dendrites that arborize extensively in superficial cortical layers, resulting in a wide horizontal distribution of dendritic contacts (Figure 3). The somatic dendrites of pyramidal cells in deep layers have a more limited spatial distribution and make predominantly local contacts with neighboring cell columns. The apical dendritic arbor, located in superficial layers, integrates relatively more global activity from a large horizontal cortical area. The somatic dendrites, in deep layers, receive relatively more local input from a smaller cortical distribution. In superficial cortical layers, activation of DA D1 receptors reportedly attenuates the propagation of a high-threshold spike (HTS) from inputs to apical dendrites. The result of attenuating the propagation of the HTS in superficial cortical layers is to diminish global activity by restricting the size of the cortical area that provides input to the pyramidal cell apical dendritic arbor.83 In deep cortical layers, however, the effect of DA D1 stimulation is to enhance an excitatory sodium conductance and attenuate an inhibitory potassium conductance, thereby potentiating the influence of the relatively more local inputs to somatic dendrites located in deeper cortical layers. With regard to the observation of reduced delta EEG activity in cocaine withdrawal,5–7,9 enhanced transmission through DA D1 receptors has been suggested to be a characteristic feature of stimulant sensitization.15,20,21,23,24,96 The apparent shift of the cortical pyramidal neuron from the global to local EEG mode via DA D1 receptor activation83 appears consistent with a hypothesis that sensitized transmission through DA D1 receptors could account for the observed deficit of delta, a forebrain global EEG mode. The direct evidence of modulatory effect of DA on membrane conductances that regulate global versus local EEG modes is strong evidence for a role of DA in the modulation of the delta rhythm and is consistent with evidence obtained from brain imaging and with the ultrastructural characteristics and laminar distribution of DA terminals.
Figure 3 places the putative generator of the delta rhythm, the pyramidal cell, in the context of some of the major modulatory influences of structures believed to be relevant to reward. According to this scheme, there are two pathways by which sensitization can reduce delta EEG power; by enhancing local mode or diminishing global mode activity of pyramidal neurons. One pathway involves the DA projections from the VTA. Enhanced signal transduction through DA D1 receptors20,21,23,24 or increased presynaptic release from VTA DA neurons14,18,24 are consistently reported features in cocaine or amphetamine sensitization, which would be expected to enhance cortical local mode activity as a consequence of DA D1 receptor stimulation of pyramidal cells.83 In addition, the context-dependent potentiation of sensitization in response to cocaine-related cues19,97,98 appears to involve descending excitatory projections from the amygdala. The descending excitatory output of the amygdala, as well as hippocampus and cortex, increases bursting of VTA DA cells, with subsequent enhancement of release from DA terminals,14,99–101 and increased local mode activity of cortical pyramidal cells.83
Another possible pathway might involve the effect of enhanced inhibition of NAc neurons as a consequence of sensitization.24–26 Following the general scheme of a striatal-pallidal-thalamic-cortical circuit,102 inhibition of NAc neurons would be expected to cause a net loss of excitatory input to the medial frontal cortex due to reduced NAc inhibitory input to medial ventral pallidum (VP), and subsequent release of inhibitory VP projections to the mediodorsal nucleus of the thalamus (MDT). The projections of the MDT to the cortex are mainly excitatory103 and directed to superficial cortical layers.82 Excitatory input from the MDT to the superficial cortical layers, which feature a wide horizontal distribution of dendrites, could be expected to enhance global EEG modes, with diminished excitatory input to the superficial cortical layers favoring a local mode response of pyramidal cells.83
POSSIBLE FUNCTIONAL SIGNIFICANCE OF REDUCED DELTA EEG POWER IN COCAINE DEPENDENCE
Reduced delta activity can be viewed as diminished resonance in the delta bandwidth. Resonance is the selective enhancement of signal transmission at specific frequencies. The delta bandwidth is an important global neocortical EEG mode that is apparently involved in the facilitation of neural signal within the neocortex and between the neocortex and subcortical structures. Evidence that delta is a neocortical resonant mode includes the observation of an association of delta frequency EEG activity with partial seizures of neocortical origin, as opposed to the association of theta range activity with allocortical foci.104 Resonance, to a significant extent, is determined by the frequency of unit activity in the underlying resonant neural structure.1,34,105 Delta-range unit activity has been observed in neocortical pyramidal neurons that are the putative generator of the surface EEG.91,93–95 Delta-range unit activity has also been recorded from subcortical structures known to play a prominent role in reward and motivation, including the NAc,41 the VP,106 and DA neurons in the VTA.14 DA-related behaviors are selectively enhanced by stimulation of DA cell axons at 2 to 3 Hz.14 Delta, a neocortical resonant mode, is a frequency bandwidth over which descending neocortical influences might relatively selectively modulate subcortical DA neurotransmission in structures relevant to reward-mediated behavior.
The functional consequence of the relative diminution of a neocortical resonant mode could involve a relative loss of neocortical modulating influence on subcortical neural transmission. Theta is a resonant mode of allocortex, as evidenced by theta frequency single-unit activity in allocortex35,107 and the apparent enhancement of neural signal transmission between allocortex and neocortex in the theta frequency bandwidth.34,35 Although theta is also apparently reduced in cocaine withdrawal, the extent of the reduction of delta is greater.5–7 The greater reduction in delta, relative to theta, would be expected to correspond to a relatively greater reduction in neocortical versus allocortical influence on subcortical neural transmission. Grace14 similarly hypothesizes a relative reduction in descending neocortical versus allocortical influence on the basis of changes in the regulation of subcortical DA neurotransmission in stimulant sensitization. As pointed out by Grace,14 the behavioral correlates of greater allocortical versus neocortical influence on subcortical activity would be expected to include features such as impulsivity, disinhibition, and affective lability that are often evident in the clinical syndrome of cocaine dependence.28
CONCLUSIONS
I have presented the argument that the abnormal power spectral profile observed in cocaine withdrawal is due to cocaine sensitization. Ultimately, for this conclusion to prove to be interesting, sensitization must be confirmed to relate importantly to the clinical syndrome of cocaine dependence, and the EEG must be found to reflect the process of sensitization with a reduction in forebrain global EEG activity. Direct evidence for sensitization and its relationship to addiction in humans would involve the finding of a progressive increase of a sign of sensitized DA transmission, such as increased DA efflux in the NAc, as attention and behavior become more exclusively focused on cocaine and its related cues. There are obvious ethical and practical limits to obtaining such information on the induction of sensitization in humans as they become dependent on cocaine. However, animal work correlating the progressive increase in signs of sensitization with cocaine-dependent behavior and EEG transition from global to local modes could clarify the significance of the apparent loss of global, and enhancement of local, EEG activity in cocaine-dependent humans.
A major difficulty in studying humans with histories of chronic cocaine use appears to be a restriction of the dynamic range of the expression of sensitization in chronic users, due to the prior induction of maximal degrees of sensitization. This effect has been evident in human studies of behavioral sensitization to cocaine or amphetamine. Human chronic cocaine users reportedly do not clearly evidence a sensitized behavioral response to the second administration of a dose of stimulant relative to the first.108,109 However, stimulant-naive subjects do evidence a sensitized behavioral response to the second of two amphetamine doses.16,17 It is apparently difficult to demonstrate the induction of sensitization with an interval change between two stimulant doses in chronic users because they may already have attained maximal degrees of expression of sensitization that no longer increment strongly with successive doses.110 Nonetheless, on the basis of evidence presented here, it is suggested that the EEG may reflect the expression of sensitization in chronic cocaine users, and that EEG might provide a noninvasive, accessible approach to investigating the possible relationship of the expression of sensitization to the clinical syndrome of cocaine dependence in human subjects.
ACKNOWLEDGMENTS
This work was supported by National Institute on Drug Abuse Grant RO1 DA077071.
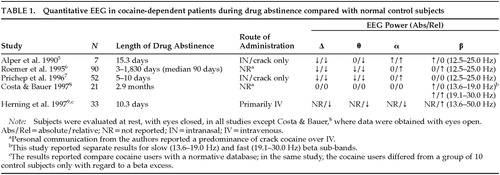 |

FIGURE 1. Transaxial views of qEEG generators visualized by using low resolution electromagnetic topographic analysis (LORETA; software from DrP. Valdes-Sosa, Cuban Neuroscience Center),80,81,111 with source superimposed on 5 slices from a probabilistic MRI atlas (software from Dr. A. C. Evans, Montreal Neurological Institute).112 The figure depicts the voxel-by-voxel analysis of variance of the Z-score relative to data obtained from 38 normal subjects for EEG power at the frequency of 2.0 Hz, across 35 individuals with crack cocaine dependence abstinent for 5 to 10 days. Color coding represents the F-value. For this comparison, the value of F adjusted for the number of variables using the correction of Worseley et al.,113 at a level of significance of P≤0.01, is marked with a white arrowhead on the color scale in the upper right corner of the figure. These data reveal a profound deficit of delta activity especially pronounced in the orbital and inferior frontal gyri, and may reflect a bias of frontal cortex towards local, as opposed to global, EEG modes because of cocaine sensitization (see text).
FIGURE 2. Schematic of horizontal integration in the frontal cortexLayer I is a “horizontal” layer that integrates limbic and association cortical input, and in turn sends axonal projections to layers II and III. Further horizontal integration across the entire vertical depth of the cortex is achieved by ascending projections of layer V–VI pyramidal cells that also terminate in layers II and III. Dopamine terminals in layers II and III are thus well positioned to selectively modulate inputs from layers I or V–VI because of the ultrastructural adaptation of the triadic complex (see text).
FIGURE 3. A putative generator of the delta EEG power, the cortical pyramidal neuron in the frontal cortex, in the context of some major regulatory relationships among structures relevant to rewardNote that the dendritic arbor of the pyramidal neuron is more horizontally extensive in upper versus lower layers. The effect of increased sensitivity of dopamine (DA) D1 receptors and activity of ventral tegmental area DA neurons, as well as enhanced inhibition of nucleus accumbens neurons occurring as a consequence of sensitization, is to diminish the influence of the more relatively global input to the pyramidal neuron dendritic arbor in the superficial layers and to potentiate the more relatively local input to somatic dendrites in the deeper layers (see text).83 Consequently, sensitization favors the transition from global to local EEG modes. Also mentioned in the text is the importance of the descending excitatory amino acid (EAA) input from the amygdala in context-dependent sensitization. Solid lines: DA; dotted lines: EAA; dashed lines: γ-aminobutyric acid (GABA).
1 Freeman WJ, Barrie JM: Chaotic oscillations and the genesis of meaning in cerebral cortex, in Temporal Coding in the Brain, edited by Buzsaki GL, Llinas R, Singer W, et al. Berlin, Springer-Verlag, 1994, pp 13–38Google Scholar
2 Alper KR, Prichep LS, Kowalik SC, et al: Persistent qEEG abnormality in crack cocaine users at 6 months of drug abstinence. Neuropsychopharmacology 1998; 19:1–9Crossref, Medline, Google Scholar
3 Volkow ND, Hitzemann R, Wang G, et al: Long-term frontal brain metabolic changes in cocaine abusers. Synapse 1992; 11:184–190Crossref, Medline, Google Scholar
4 Johanson C-E, Schuster CR: Cocaine, in Psychopharmacology: The Fourth Generation of Progress, edited by Bloom FE, Kupfer DJ. New York, Raven, 1995, pp 1685–1697Google Scholar
5 Alper KR, Chabot RJ, Kim AH, et al: Quantitative EEG correlates of crack cocaine dependence. Psychiatry Res 1990; 35:95–106Crossref, Medline, Google Scholar
6 Roemer RA, Cornwall A, Dewart D, et al: Quantitative electroencephalographic analysis in cocaine-preferring polysubstance abusers during abstinence. Psychiatry Res 1995; 58:247–257Crossref, Medline, Google Scholar
7 Prichep LS, Alper KR, Kowalik SC, et al: Quantitative electroencephalographic characteristics of crack cocaine dependence. Biol Psychiatry 1996; 40:986–993Crossref, Medline, Google Scholar
8 Costa L, Bauer L: Quantitative electroencephalographic differences associated with alcohol, cocaine, heroin and dual-substance dependence. Drug Alcohol Depend 1997; 46:87–93Crossref, Medline, Google Scholar
9 Herning RI, Guo X, Better WE, et al: Neurophysiological signs of cocaine dependence: increased electroencephalogram beta during withdrawal. Biol Psychiatry 1997; 41:1087–1094Google Scholar
10 Prichep LS, Alper KR, Kowalik S, et al: Prediction of treatment outcome in cocaine dependent males using EEG. Drug Alcohol Depend 1999; 54:35–43Crossref, Medline, Google Scholar
11 Alper K: Quantitative EEG and evoked potentials in adult psychiatry, in Advances in Biological Psychiatry, vol 1, edited by Panksepp J. Greenwich, CT, JAI Press, 1995, pp 65–112Google Scholar
12 Chabot RJ, Serfontein G: Quantitative EEG profiles of children with attention deficit disorder. Biol Psychiatry 1996; 40:951–963Crossref, Medline, Google Scholar
13 Kalivas PW, Sorg BA, Hooks MS: The pharmacology and neural circuitry of sensitization to psychostimulants. Behav Pharmacol 1993; 4:315–334Crossref, Medline, Google Scholar
14 Grace AA: The tonic/phasic model of dopamine system regulation: its relevance for understanding how stimulant abuse can alter basal ganglia function. Drug Alcohol Depend 1995; 37:111–129Crossref, Medline, Google Scholar
15 White FJ, Kalivas PW: Neuroadaptations involved in amphetamine and cocaine addiction. Drug Alcohol Depend 1998; 51:141–153Crossref, Medline, Google Scholar
16 Strakowski SM, Sax KW, Setters MJ, et al: Enhanced response to repeated d-amphetamine challenge: evidence for behavioral sensitization in humans. Biol Psychiatry 1996; 40:872–880Crossref, Medline, Google Scholar
17 Gorelick DA, Rothman DB: Stimulant sensitization in humans. Biol Psychiatry 1997; 40:230–231Crossref, Google Scholar
18 Kalivas PW, Churchhill L, Klitenick MA: The circuitry mediating the translation motor responses motivational stimuli into adaptive motor responses, in Limbic Motor Circuits and Neuropsychiatry, edited by Kalivas PW, Barnes CD. Boca Raton, FL, CRC Press, 1993, pp 239–309Google Scholar
19 Anagnostaras SG, Robinson TE: Sensitization to the psychomotor stimulant effects of amphetamine: modulation by associative learning. Behav Neurosci 1996; 110:1397–1414Google Scholar
20 Bonci A, Williams JT: A common mechanism mediates long-term changes in synaptic transmission after chronic cocaine and morphine. Neuron 1996; 16:631–639Crossref, Medline, Google Scholar
21 Pierce RC, Born B, Adams M, et al: Repeated intra-ventral tegmental area administration of SKF-38393 induces behavioral and neurochemical sensitization to a subsequent cocaine challenge. J. Pharmacol Exp Ther 1996; 8:384–392Google Scholar
22 White FJ: Cocaine actions in mesocorticolimbic dopamine systems: role in sensitization and withdrawal, in Problems of Drug Dependence: Proceedings of the 57th Annual Scientific Meeting, Research Monograph No. 162, edited by Harris LS. Rockville, MD, National Institute on Drug Abuse, 1996, pp 76–77Google Scholar
23 Kelley AE, Holahan MR: Enhanced reward-related responding following cholera toxin infusion into the nucleus accumbens. Synapse 1997; 26:46–54Crossref, Medline, Google Scholar
24 Henry DJ, White FJ: The persistence of behavioral sensitization to cocaine parallels enhanced inhibition of nucleus accumbens neurons. J. Neurosci 1995; 15:6287–6299Google Scholar
25 Kiyatkin EA, Rebec GV: Dopaminergic modulation of glutamate-induced excitations of neurons in the neostriatum and nucleus accumbens of awake, unrestrained rats. J Neurophysiol 1996; 75:142–153Crossref, Medline, Google Scholar
26 O'Donnell P, Grace AA: Dopaminergic reduction of excitability in nucleus accumbens neurons recorded in vitro. Neuropsychopharmacology 1996; 15:87–97Crossref, Medline, Google Scholar
27 Heyman GM: Resolving the contradictions of addiction. Behav Brain Sci 1996; 19:561–610Crossref, Google Scholar
28 Robinson TE, Berridge KC: The neural basis of drug craving: an incentive-sensitization theory of addiction. Brain Res Rev 1993; 18:247–291Crossref, Medline, Google Scholar
29 Kiyatkin EA: Functional significance of mesolimbic dopamine. Neurosci Biobehav Rev 1995; 19:578–598Google Scholar
30 Salamone JD, Cousins MS, Snyder BJ: Behavioral functions of nucleus accumbens dopamine: empirical and conceptual problems with the anhedonia hypothesis. Neurosci Biobehav Rev 1997; 21:341–359Crossref, Medline, Google Scholar
31 Pülvermuller F, Birbaumer N, Lutzenberger W, et al: High-frequency brain activity: its possible role in attention, perception and language processing. Prog Neurobiol 1997; 52:427–445Crossref, Medline, Google Scholar
32 Self DW, Nestler EJ: Relapse to drug seeking: neural and molecular mechanisms. Drug Alcohol Depend 1998; 51:49–60Crossref, Medline, Google Scholar
33 Nunez PL: Experimental connections between EEG data and the global wave theory, in Neocortical Dynamics and Human EEG Rhythms, edited by Nunez PL. New York, Oxford University Press, 1995, pp 534–590Google Scholar
34 Steriade M, Gloor P, Llinas RR, et al: Basic mechanisms of cerebral rhythmic activities. EEG Clin Neurophysiol 1990; 76:481–508Crossref, Medline, Google Scholar
35 Miller R: Cortico-Hippocampal Interplay and the Representation of Contexts in the Brain. Berlin, Springer-Verlag, 1991Google Scholar
36 Luoh H-F, Kuo TB, Chan AH, et al: Power spectral analysis of electroencephalographic desynchronization induced by cocaine in rats: correlation with microdialysis evaluation of dopaminergic neurotransmission at the medial prefrontal cortex. Synapse 1994; 16:29–35Crossref, Medline, Google Scholar
37 Chang AW, Kuo TJ, Chen CF, et al: Power spectral analysis of electroencephalographic desynchronization induced by cocaine in rats: correlation with evaluation of noradrenergic neurotransmission at the medial prefrontal cortex. Synapse 1995; 21:149–157Crossref, Medline, Google Scholar
38 Kropf W, Kuschinsky K: Effects of stimulation of Dopamine D1 receptors on the cortical EEG in rats: different influences by a blockade D2 receptors and by an activation of putative dopamine autoreceptors. Neuropharmacology 1993; 32:493–500Crossref, Medline, Google Scholar
39 Ferger B, Kropf W, Kuschinsky K: Studies on electroencephalogram (EEG) in rats suggest that moderate doses of cocaine or d-amphetamine activate D1 rather than D2 receptors. Psychopharmacology 1994; 114:297–308Crossref, Medline, Google Scholar
40 Ferger B, Stahl D, Kuschinsky K: Effects of cocaine on the EEG power spectrum of rats are significantly altered after its repeated administration: do they reflect sensitization phenomena? Naunyn Schmiedebergs Arch Pharmacol 1996; 353:545–551Google Scholar
41 Leung LS, Yim CY: Rhythmic delta-frequency activities in the nucleus accumbens of anesthetized and freely moving rats. Can J Physiol Pharmacol 1993; 71:311–320Crossref, Medline, Google Scholar
42 Herning RI, Jones RT, Hooker WD, et al: Cocaine increases EEG beta: a replication and extension of Hans Berger's historic experiments. EEG Clin Neurophysiol 1985; 60:470–477Crossref, Medline, Google Scholar
43 Mannelli P, Janiri L, Tempesta E, et al: Prediction in drug abuse: cocaine interactions with alcohol and buprenorphine. Br J Psychiatry 1993; 163(21, suppl):39–45Google Scholar
44 Herning RI, Glover BJ, Koeppl B, et al: Cocaine-induced increases in EEG alpha and beta activity: evidence for reduced cortical processing. Neuropsychopharmacology 1994; 11:1–9Crossref, Medline, Google Scholar
45 Lukas SE: Topographic brain mapping during cocaine-induced intoxication and self-administration, in Biological Psychiatry, vol 2, edited by Racagni G, Brunello N, Fukuda T. Amsterdam, Elsevier Science, 1991, pp 25–29Google Scholar
46 Noldy NE, Santos CV, Politzer N, et al: Quantitative EEG changes in cocaine withdrawal: evidence for long-term CNS effects. Neuropsychobiology 1994; 30:189–196Crossref, Medline, Google Scholar
47 Bauer LO: Electroencephalographic and autonomic predictors of relapse in alcohol-dependent patients. Alcohol Clin Exp Res 1994; 18:755–760Crossref, Medline, Google Scholar
48 Winterer G, Koppel B, Heinz A, et al: Quantitative EEG (QEEG) predicts relapse in patients with chronic alcoholism and points to a frontality pronounced cerebral disturbance. Psychiatry Res 1998; 78:101–113Crossref, Medline, Google Scholar
49 Rougeul-Buser A: Electrocortical rhythms in the 40 Hz band in cat: in search of their behavioral correlates, in Temporal Coding in the Brain, edited by Buzsaki G, Llinas R, Singer W, et al. Berlin, Springer-Verlag, 1994, pp 103–114Google Scholar
50 Struve FA, Straumanis JJ, Patrick G, et al: Topographic mapping of quantitative EEG variables in chronic heavy marijuana users: empirical findings with psychiatric patients. Clin Electroencephalogr 1989; 20:6–23Crossref, Medline, Google Scholar
51 Struve FA, Straumanis JJ, Patrick G: Persistent topographic quantitative EEG sequelae of chronic marijuana use: a replication study and initial discriminant function analysis. Clin Electroencephalogr 1994; 25:63–75Crossref, Medline, Google Scholar
52 Riedel R-R, Alper KR, Bulau P, et al: QEEG in hemophiliacs with HIV infection. Clin Electroencephalogr 1995; 26:84–91Crossref, Medline, Google Scholar
53 Matousěk M, Petersén I: Norms for the EEG, in Automation of Clinical Electroencephalography, edited by Kellaway P, Petersén, I. New York, Raven, 1973, pp 75–102Google Scholar
54 Gasser T, Bacher P, Mochs J: Transformation towards the normal distribution of broadband spectral parameters of the EEG. Electroencephalogr Clin Neurophysiol 1982; 53:119–124Crossref, Medline, Google Scholar
55 Jonkman EJ, Poortvliet DCJ, Veering MM, et al: The use of neurometrics in the study of patients with cerebral ischemia. Electroencephalogr Clin Neurophysiol 1985; 61:333–341Crossref, Medline, Google Scholar
56 Alvarez A, Pascual R, Valdes P: U.S. EEG developmental equations confirmed for Cuban schoolchildren. Electroencephalogr Clin Neurophysiol 1987; 67:330–332Crossref, Medline, Google Scholar
57 Harmony T, Alvarez A, Pascual R, et al: EEG maturation of children with different economic and psychosocial characteristics. Int J Neurosci 1987; 31:103–113Google Scholar
58 John ER, Prichep LS, Easton P: Normative data banks and neurometrics: basic concepts, methods and results of norm construction, in Handbook of Electroencephalography and Clinical Neurophysiology, vol III, edited by Remond A. Amsterdam, Elsevier, 1987, pp 449–495Google Scholar
59 Harmony T, Fernández T, Silva J, et al: EEG delta activity: an indicator of attention to internal processing during performance of mental tasks. Int J Psychophysiol 1996; 24:161–171Crossref, Medline, Google Scholar
60 Fernandez T, Harmony T, Rodriguez M, et al: EEG activation patterns during the performance of tasks involving different components of mental calculation. Electroencephalogr Clin Neurophysiol 1995; 94:175–182Crossref, Medline, Google Scholar
61 Van Dijk JG, Caekebeke JFV, Jennekens Schinkel A, et al: Background EEG reactivity in auditory event-related potentials. Electroencephalogr Clin Neurophysiol 1992; 83:44–51Crossref, Medline, Google Scholar
62 Michel CM, Henggeler B, Brandeis D, et al: Localization of sources of brain alpha/theta/delta activity and the influence of the mode of spontaneous mentation. Physiol Meas 1993; 14:21–26Crossref, Google Scholar
63 Basar-Eroglu C, Basar E, Demiralp T, et al: P300-response: possible psychophysiological correlates in delta and theta frequency channels: a review. Int J Psychophysiol 1992; 13:161–179Crossref, Medline, Google Scholar
64 Roschke J, Fell J: Spectral analysis of P300 generation in depression and schizophrenia. Neuropsychobiology 1997; 35:108–114Crossref, Medline, Google Scholar
65 John ER, Easton P, Isenhart R, et al: Electrophysiological analysis of the registration, storage and retrieval of information in delayed match from sample. Int J Psychophysiol 1996; 24:127–144Crossref, Medline, Google Scholar
66 Bullock TH: Introduction to induced rhythms: a widespread, heterogeneous class of oscillations, in Induced Rhythms in the Brain, edited by Basar E, Bullock TH. Boston, Birkhauser, 1992, pp 1–26Google Scholar
67 Guido W, Weyand T: Burst responses in thalamic relay cells of the awake behaving cat. J Neurophysiol 1995; 74:1782–1786Google Scholar
68 Biederman J, Wilens T, Mick E, et al: Psychoactive substance use disorders in adults with attention deficit hyperactivity disorder (ADHD): effects of ADHD and psychiatric comorbidity. Am J Psychiatry 1995; 152:1652–1658Google Scholar
69 Regan D: Human Brain Electrophysiology. Amsterdam, Elsevier, 1989Google Scholar
70 Basar E, Basar-Eroglu C, Rosen B, et al: A new approach to endogenous event related potentials in man: relation between EEG and P300 wave. Int J Neurosci 1984; 26:161–180Crossref, Google Scholar
71 Intriligator J, Polich J: On the relationship between EEG and ERP variability. Int J Psychophysiol 1995; 20:59–74Crossref, Medline, Google Scholar
72 Michel CM, Lehmann D, Henggeler B, et al: Localization of the sources EEG delta, theta, alpha and beta frequency bands using the FFT dipole approximation. Electroencephalogr Clin Neurophysiol 1992; 82:38–44Crossref, Medline, Google Scholar
73 Alper K, Günther W, Prichep LS, et al: Correlation of qEEG with PET in schizophrenia. Neuropsychobiology 1998; 38:50–56Crossref, Medline, Google Scholar
74 Alper KR, Prichep LS, John ER, et al: QEEG in cocaine withdrawal: possible neurophysiological implications regarding the generation of slow EEG activity. 6th International Congress of the International Society for Brain Electromagnetic Topography (ISBET), Tokushima, Japan, 1995, Abstract No. S-2-GGoogle Scholar
75 Maquet P, Degueldre C, Delfiore G, et al: Functional neuroanatomy of human slow wave sleep. J Neurosci 1997; 17:2807–2812Google Scholar
76 Hofle N, Paus T, Reutens D, et al: Regional cerebral blood flow changes as a function of delta and spindle activity during slow wave sleep in humans. J Neurosci 1997; 17:4800–4808Google Scholar
77 Gatley SJ, Volkow ND: Addiction and imaging of the living human brain. Drug Alcohol Depend 1998; 51:97–108Crossref, Medline, Google Scholar
78 Strickland TL, Villanueva-Meyer J, Miller BL, et al: Cerebral perfusion and neuropsychological consequences of chronic cocaine use. J Neuropsychiatry Clin Neurosci 1993; 5:419–427Link, Google Scholar
79 Levin JM, Holman BL, Mendelson JH, et al: Gender differences in cerebral perfusion in cocaine abuse. J Nucl Med 1994; 35:1902–1909Google Scholar
80 Pascual-Marqui RD, Michel CM, Lehmann D: Low resolution electromagnetic tomography: a new method for localizing electrical activity in the brain. Int J Psychophysiol 1994; 18:49–65Crossref, Medline, Google Scholar
81 Lantz G, Michel CM, Pascual-Marqui RD, et al: Extracranial localization of intracranial interictal epileptiform activity using LORETA (low resolution electromagnetic tomography). Electroencephalogr Clin Neurophysiol 1997; 102:414–422Crossref, Medline, Google Scholar
82 Barbas H: Anatomic basis of cognitive-emotional interactions in the primate prefrontal cortex. Neurosci Biobehav Rev 1995; 19:499–510Crossref, Medline, Google Scholar
83 Yang CR, Seamans JK: Dopamine D1 receptor actions in layers V–VI rat prefrontal cortex neurons in vitro: modulation of dendritic-somatic signal integration. J Neurosci 1996; 16:1922–1935Google Scholar
84 Berger B: Distinctive chemoanatomical and developmental features of the prefrontal dopaminergic system in primates as compared to rodents, in Motor and Cognitive Functions of the Prefrontal Cortex, edited by Thierry AM, Glowinski PS, Goldman-Rakic PS, et al. Berlin and Heidelberg, Springer-Verlag, 1994, pp 17–34Google Scholar
85 Hestrin S, Armstrong WE: Morphology and physiology of cortical neurons in layer I. J Neurosci 1996; 16:5290–5300Google Scholar
86 Nunez PL: Mind, brain and electroencephalography, in Neorcortical Dynamics and Human EEG Rhythms, edited by ñez PL. New York, Oxford University Press, 1995, pp 133–194Google Scholar
87 Silberstein RB: Neuromodualation and neocortical dynamics, in Neocortical Dynamics and Human EEG Rhythms, edited by Nunez PL. New York, Oxford University Press, 1995, pp 591–627Google Scholar
88 Verney C, Alvarez C, Geffard M, et al: Ultrastructural double labeling study of dopamine terminals and GABA-containing neurons in rat anteromedial cerebral cortex. Eur J Neurosci 1990; 2:960–972Crossref, Medline, Google Scholar
89 Camp DM, DeJonghe DK, Robinson TE: Time-dependent effects of repeated amphetamine treatment on norepinephrine in the hypothalamus and hippocampus assessed with in vivo microdialysis. Neuropsychopharmacology 1997; 17:130–140Crossref, Medline, Google Scholar
90 Anthony JC, Tien AY, Petronis KR: Epidemiologic evidence on cocaine use and panic attacks. Am J Epidemiol 1989; 129:543–549Crossref, Medline, Google Scholar
91 Hutcheon B, Miura RM, Puil E: Subthreshold membrane resonance in neocortical neurons. J Neurophysiol 1996; 76:683–697Crossref, Medline, Google Scholar
92 Yang CR, Seamans JK, Gorelova N: Electrophysiological and morphological properties of layers V–VI principal pyramidal cells in rat prefrontal cortex in vitro. J Neurosci 1996; 16:1904–1921Google Scholar
93 Steriade M, Nuñez A, Amzica F: A novel slow (<1 Hz) oscillation of neocortical neurons in vivo: depolarizing and hyperpolarizing components. J Neurosci 1993; 13:3252–3265Google Scholar
94 Steriade M, Nuñez A, Amzica F: Intracellular analysis of relations between the slow (<1 Hz) neocortical oscillation and other sleep rhythms of the electroencephalogram. J Neurosci 1993; 13:3266–3283Google Scholar
95 Steriade M, Nuñez A, Amzica F: The slow (<1 Hz) oscillation in reticular thalamic and thalamocortical neurons: scenario of sleep rhythm generation in interacting thalamic and neocortical networks. J Neurosci 1993; 13:3284–3299Google Scholar
96 Unterwald EM, Fillmore J, Kreek MJ: Chronic repeated cocaine administration increases dopamine D1 receptor-mediated signal transduction. Eur J Pharmacol 1996; 318:31–35Crossref, Medline, Google Scholar
97 Burechailo L, Martin-Iverson MT: Behavioral sensitization to cocaine, but not cocaine-conditioned behavior, is associated with increased dopamine occupation of its receptors in the nucleus accumbens. Behav Neurosci 1996; 110:1388–1396Google Scholar
98 Crombag HS, Badiani A, Robinson TE: Signalled versus unsignalled intravenous amphetamine: large differences in the acute psychomotor response and sensitization. Brain Res 1996; 25:227–231Crossref, Google Scholar
99 Karreman M, Moghaddam B: The prefrontal cortex regulates the basal release of dopamine in the limbic striatum: an effect mediated by ventral tegmental area. J Neurochem 1996; 66:589–598Crossref, Medline, Google Scholar
100 Brudzynski SM, Gibson CJ: Release of dopamine in the nucleus accumbens caused by stimulation of the subiculum in freely moving rats. Brain Res Bull 1997; 42:303–308Crossref, Medline, Google Scholar
101 Taber MT, Fibiger HC: Feeding-evoked dopamine release in the nucleus accumbens: regulation by glutamatergic mechanisms. Neuroscience 1997; 76:1105–1112Google Scholar
102 Mogenson GJ, Brudzynski SM, Wu M, et al: From motivation to action: a review of dopaminergic regulation of limbic → nucleus accumbens → ventral pallidum → pedunculopontine nucleus circuitries involved in limbic-motor integration, in Limbic Motor Circuits and Neuropsychiatry, edited by Kalivas PW, Barnes CD. Boca Raton, FL, CRC Press, 1993, pp 193–236Google Scholar
103 Jones MW, Kilpatrick IC, Phillipson OT: Regulation of dopamine function in the prefrontal cortex of the rat by the thalamic mediodorsal nucleus. Brain Res Bull 1987; 19:9–17Crossref, Medline, Google Scholar
104 Ebersole JS, Pacia SV: Localization of temporal lobe foci by ictal EEG patterns. Epilepsia 1996; 37:386–399Crossref, Medline, Google Scholar
105 Creutzfeldt OD, Ojemann GA, Chatrian GE: Activity of single neurons and their relationship to normal EEG waves and interictal epilepsy potentials in humans, in Slow Potential Changes in the Brain, edited by Haschke W, Speckman EJ, Roitbak AI. Boston, Birkhauser, 1993, pp 21–42Google Scholar
106 Lavin A, Grace AA: Physiological properties of rat ventral pallidal neurons recorded intracellularly in vivo. J Neurophysiol 1996; 75:1432–1443Google Scholar
107 Skaggs W, McNaughton BL, Wilson MA, et al: Theta phase procession in hippocampal neuronal populations and the compression of temporal sequences. Hippocampus 1996; 6:149–172Crossref, Medline, Google Scholar
108 Rothman RB, Gorelick GA, Guo XY, et al: Lack of evidence for context specific cocaine induced sensitization in humans: preliminary studies. Pharmacol Biochem Behav 1994; 49:583–588Crossref, Medline, Google Scholar
109 Rothman RB, Henningfield JE, Johanson CE, et al: Failure to detect cocaine-induced context-specific sensitization in human cocaine addicts, in Problems of Drug Dependence, 1996: Proceedings of the 58th Annual Scientific Meeting, The College on Problems of Drug Dependence Inc., Research Monograph No. 174, edited by Harris LS. Rockville, MD, National Institute on Drug Abuse, 1977, p 177Google Scholar
110 Phillips TJ: Behavioral genetics of drug sensitization. Critical Reviews in Neurobiology 1997; 11:21–33Crossref, Medline, Google Scholar
111 Valdes-Sosa P: Quantitative electroencephalographic tomography (abstract). Electroencephalogr Clin Neurophysiol 1997; 103:19Crossref, Google Scholar
112 Evans AC, Collins DL, Mills SR, et al:3D statistical neuroanatomical models from 305 MRI volumes, in Proceedings of IEEE-Nuclear Science Symposium and Medical Imaging Conference, 1993, pp 1813–1817Google Scholar
113 Worseley KJ, Marrett S, Neelin P, et al: A unified statistical approach for determining significant signals in images of cerebral activation. Human Brain Mapping 1996; 4:58–73Crossref, Medline, Google Scholar

