
Clinicopathologic Case Report
PRESENTATION OF THE CASE
A 57-year-old right-handed man presented with an 18-month history of progressive cognitive impairment leading to immobility and mutism. The patient initially developed memory problems and gait instability. Six months after the onset of these symptoms, he was evaluated by a physician who described the patient as withdrawn and unable to follow complex commands. Three months after this doctor visit, the patient had the onset of generalized tonic-clonic seizures, requiring multiple anticonvulsant regimens and several hospitalizations before obtaining seizure control. Subsequently he continued to have decreased spontaneous behaviors, progressive mental slowing, disorientation to time, and poor memory.
The patient was of Guamanian Chamorro origin, had a 12th-grade education, and worked as a paralegal. His family history was significant for an older sister with Parkinson's disease and mild dementia, which was not felt to be due to the parkinsonism dementia complex of Guam. There was no family history of cerebrovascular diseases. His past medical history was significant for previously treated hypertension, but there was no history of stroke, diabetes, cardiovascular disease, migraine headaches, alcoholism, or toxic exposures. On presentation, he was taking valproate (200 mg q6h) and phenobarbital (60 mg bid) but no longer required antihypertensive medications.
The patient's examination was remarkable for an akinetic-mute state. Vital signs were within normal limits, except for elevated blood pressures in the 153/97 range. His eyes were open and he appeared awake, but he did not initiate any motor or verbal behavior. He blinked to visual threat and visually tracked people and environmental movement. On attempted interview, the patient looked directly at the examiner but did not respond to any verbal commands, although he withdrew all extremities from noxious stimuli. The patient remained bedridden and incontinent, had to be fed, and had a normal wake-sleep cycle. His pupils were equal and reactive to light, oculocephalic and corneal reflexes were present, and bilateral gag reflexes appeared normal and symmetric. The motor evaluation showed increased tone of a spastic “clasp-knife” nature rather than an extrapyramidal rigidity. There was no myoclonus or other movement disorder. Deep tendon reflexes were within normal limits, but plantar responses were upgoing. He had positive glabellar and snout reflexes.
The patient underwent an evaluation for an akinetic-mute state associated with a rapidly progressive dementing illness and generalized seizures. All routine laboratory results were normal, including blood cell counts, metabolic panel, renal and hepatic tests, and inflammatory and autoimmune screen. Human immunodeficiency virus serology and blood and urine cultures were negative. An electroencephalogram (EEG) disclosed generalized slowing without evidence of seizure activity or of periodic discharges. Magnetic resonance imaging (MRI) of the brain showed extensive and diffuse signal abnormalities in a bilaterally symmetric fashion involving the subcortical white matter of both hemispheres (Figures 1, 2, and 3). There was generalized cerebral atrophy and thinning of the corpus callosum. Magnetic resonance angiography did not show changes consistent with a vasculitis, and single-photon emission computed tomography (SPECT) disclosed diffuse, widespread hypoperfusion. The cerebrospinal fluid (CSF) had normal glucose and cell counts with a mildly elevated protein level (67 mg/dl). CSF syphilis serology was nonreactive, and bacterial, viral, and fungal cultures were negative. CSF immunoglobulin and γ-immunoglobulin (IgG) synthesis levels were also normal, and there were no oligoclonal bands. In addition, the patient's CSF was negative for the 14-3-3 protein marker associated with Creutzfeldt-Jakob disease (CJD).1
The patient's clinical course continued to be one of relentless deterioration. He remained in an akinetic mute state without any spontaneous behavior, dependent for all activities of daily living, and fed through a percutaneous endoscopic gastrostomy tube. He died within a year of his assessment from a cardiorespiratory arrest.
CLINICAL DISCUSSION
The patient presented with a rapidly progressive dementia with akinetic mutism, seizures, and upper motor neuron signs. In addition, his neuroimaging disclosed diffuse white matter disease. This pattern of deficits raised several concerns in the differential diagnosis.
The main manifestation of this patient's illness was akinetic mutism. Patients with akinetic mutism appear cognizant of the activity around them and are able to move and speak, but do not. Lesions that produce the commonest form of akinetic mutism, also called coma vigil, involve bilateral medial frontal areas, particularly the anterior cingulate gyrus or its projections.2 Akinetic mutism may also result from lesions of the ventral striatum or globus pallidus, the medial thalamus, the medial forebrain bundle as it courses through the anterior hypothalamus, and the midbrain reticular structures or their course through the subthalamic region. Specific causes include frontal trauma, bilateral anterior cerebral artery occlusions, hydrocephalus, bilateral thalamic infarction, diencephalic area tumors, subthalamic or midbrain strokes, and advanced frontal-subcortical dementing illnesses.
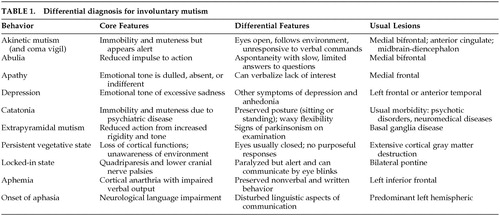
Clinicians must distinguish akinetic mutism from other potential causes of involuntary mutism (Table 1). Akinetic mutism differs from the milder abulia, which is characterized by a reduced impulse to act, slow and delayed responses, and difficulty performing more than one step in a series. Apathy and depression are alterations in emotional tone rather than action or motor initiation. Catatonia is marked by associated features of waxy flexibility, catatonic posturing, or mannerisms. Extrapyramidal mutism may result from marked parkinsonian rigidity. Patients with the persistent vegetative state or apallic state have lost cognitive abilities but retain basic brainstem functions such as respiratory and cardiovascular regulation. In the locked-in syndrome, basilar artery occlusion with pontine infarct causes a state of motor paralysis where only ocular movements remain. Aphemia and the onset of several aphasia syndromes include an inability to produce verbal output, but with intact nonverbal behavior.
Two early etiologic considerations in this patient were Creutzfeldt-Jakob disease (CJD) and the parkinsonism dementia complex (PDC) of Guam.3,4 The rapid progression of the clinical dementia dictated consideration of CJD, a subacute prion encephalopathy associated with myoclonus and gait difficulty.3 Our patient, however, lacked supportive findings for CJD, such as 1–2 cycles/second triphasic sharp complexes on EEG and the 14-3-3 protein marker for CJD in the CSF.1 The PDC of Guam, which is often associated with amyotrophic lateral sclerosis, is a neurodegenerative disorder endemic to the Chamorros of Guam.4 The patient's rapid clinical course, lack of parkinsonism or motor neuron disease, and extensive white matter involvement made the diagnosis of PDC unlikely.
The differential diagnosis focused on the presence of extensive white matter hyperintensities (WMH) on MRI (Table 2).5 Periventricular white matter abnormalities, or leukoaraiosis, consist of ill-defined regions with low attenuation values around the frontal and occipital horns of the lateral ventricles, often extending into the white matter of the centrum semiovale.6,7 Some degree of leukoaraiosis occurs in the majority of individuals over the age of 75, and many of these individuals are cognitively intact.7–10 Mild leukoaraiosis with smooth periventricular rims and thin caps may be nonischemic, from breakdown of the ependyma and subependymal gliosis, but severe leukoaraiosis with extensive periventricular rims and patchy and confluent deep white matter hyperintensities typically results from demyelinating disease or ischemic injury.7 The absence of CSF IgG changes consistent with demyelinating disease suggested that ischemic injury was the more likely explanation. Periventricular and deep white matter are perfused by distal branches of penetrating arterioles that are susceptible to chronic ischemia,11,12 which results in arteriolosclerosis with white matter degeneration, demyelination, reactive gliosis, hyalinosis, and fibrinoid necrosis.7,13
This patient's WMH may represent a form of vascular dementia (VaD) from leukoaraiosis.14,15 The extent of these lesions on T2-weighted MR images can be an indicator of cognitive impairment,10,16–20 and extensive leukoaraiosis is more common in demented patients than in nondemented ones.10,12,19,21,22 Callosal atrophy, present in our patient, may be another important predictor of global cognitive impairment in patients with WMH.23 The presence of severe leukoaraiosis may be associated with decreased psychomotor speed and mental control, abnormal sustained and divided attention, memory retrieval difficulty, decreased performance IQ, frontal executive dysfunction, and decreased flexibility of mental functions.22,24–26 The preferential subfrontal location of the white matter lesions suggests that involvement of subcortical-frontal behavioral circuits and cingulate involvement are responsible for these neurobehavioral changes.23,27
VaD with extensive WMH can have several causes. Some of these are familial, such as cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL),28 Fabry's disease,29 and other rare hereditary cerebrovascular disorders.30,31 Gradually progressive dementia without discrete cerebrovascular events can result in Sneddon's syndrome in a patient with widespread livido reticularis.32 When hereditary and other specific causes are excluded, we are left with severe leukoaraiosis, otherwise known as Binswanger's disease.33,34 Otto Binswanger first described “subcortical arteriosclerotic encephalopathy” as a prominent dementia with a history of hypertension and extensive periventricular white matter rarefaction on pathology.35 As in our patient, the victims of Binswanger's disease have gradually progressive aspontaneity, slowed cognition, frequent falls, upper motor neuron signs, and possible seizures.36
Unusual features for Binswanger's disease in our patient are the rapidity of the course and the absence of severe hypertension at the time of presentation. Although the typical course of Binswanger's disease is about five years, there is a wide range of durations, and this patient's eventual 2- to 3-year course is still compatible with this diagnosis. Furthermore, the clinical diagnosis of Binswanger's disease demands a history of chronic hypertension rather than evidence of markedly elevated blood pressures at the time of assessment. Finally, the definitive diagnosis of Binswanger's disease requires pathological confirmation. Early in the course, SPECT scans may show decreased perfusion in frontal cortex and basal ganglia,35 but late in the course, as in this patient, the hypoperfusion is more diffuse and nonspecific. In the future, newer laboratory findings may support the diagnosis of Binswanger's disease during life, such as magnetization transfer images,37 ischemia on diffusion-weighted MRI,38 slowed arterial-venous transit times by transcranial sonography,39 and decreased cerebral reactivity to apnea during cognitive tasks.40
CLINICAL DIAGNOSIS
Binswanger's disease.
PATHOLOGIC DISCUSSION
The gross neuropathology was primarily remarkable for white matter changes. The brain weighed 1,210 grams prior to fixation, and there was no evidence of hemorrhage, mass effect, or cerebral edema. The cerebral hemispheres were symmetrical, were uniformly cream-colored, and had a normal pattern of sulci and gyri. On serial sections (coronal plane at approximately l cm intervals), the cortical ribbon appeared intact, without cysts or infarcts. The telencephalic deep gray structures and the hippocampi were unremarkable, as was most of the neocortex. In contrast, the subcortical white matter was diffusely softened, with a “moth-eaten” appearance (Figure 4), a change most prominent in the frontal lobes, with corresponding dilatation of the lateral and third ventricles. Other observations included mild atherosclerosis of the basal vessels and normal examinations of the brainstem, including substantia nigra and locus ceruleus, and the cerebellum.
On histological examination, there were multiple patchy regions of pallor with prominent demyelination of the subcortical white matter (Figure 5 A, B). There was a profound arteriosclerotic changes in deep penetrating end-arterioles, especially in periventricular and deep white matter, characterized by the replacement of smooth muscle fibers by fibrohyaline material (Figure 6). These changes were associated with focally prominent adventitial inflammation composed primarily of scattered lymphocytes in the vascular adventitia and throughout the white matter (Figure 6). In addition, there were prominent microglia in both white matter and adjacent cortex and scattered collections of macrophages present in white matter adjacent to the deep white matter, deep central gray matter, and midbrain.
In general, this patient did not show lacunar infarctions, but rather a severe multifocal patchy rarefaction of the white matter with collections of histiocytes, especially prominent around the blood vessels. These vascular changes were also present, though to a lesser degree, in the basal ganglia and the cortex. There was no evidence of neurofibrillary tangles, diffuse neuritic plaques or granulovacuolar degeneration, Lewy bodies, or amyloid angiopathy in the entorhinal cortex, hippocampus, or neocortex. There were no spongiform changes in the neuropil of the cortex and no viral inclusions within the nuclei or cytoplasm of any neuroglial cells. There was severe neuron loss and gliosis, however, in the hippocampus (especially the CA1 sector) consistent with severe unilateral hippocampal sclerosis. Finally, there was no evidence of granular osmiophilic material deposition or of specific changes seen with CADASIL or other genetic cerebrovascular diseases.28
Recent work demonstrates astrocytic gliosis, activation of microglia, and an inflammatory response in clinicopathologically proven Binswanger's disease.41 The pathological alterations include a decrease in the oligodendroglia, which are associated with the degradation of both myelin and axonal components.41 Immunohistochemical characterization of tissue sections was carried out by using primary antibodies to glial fibrillary acidic protein (GFAP, an astrocyte marker), CD68 (a macrophage/microglial marker), and CD3 (a T-cell marker), all with appropriate positive and negative controls. GFAP immunohistochemistry showed astrocytic gliosis in the subcortical white matter but a surprisingly severe degree of astrogliosis in the overlying neocortex (Figure 7 A). CD68 immunohistochemistry showed prominent macrophages in thickened arterial walls, as well as abundant, evenly spaced microglia and macrophages in the adjacent subcortical white matter (Figure 7 B). Anti-CD3 immunostaining also disclosed immunoreactive cells (Figure 7 C). These results were consistent with the hypothesis that an inflammatory reaction may play an important role in the pathophysiology of Binswanger's disease.41
In sum, the patchy subcortical leukoencephalopathy associated with severe arteriosclerotic change findings in this patient are diagnostic of Binswanger's disease.42–44 This patient was unique in having extreme Binswanger's disease without accompanying lacunes or cystic lesions, as are usually seen in the presence of hypertensive cerebrovascular arteriolosclerosis. The other unusual feature of this patient was the focally severe degree of adventitial inflammation, including macrophages and T lymphocytes, around the white matter microvessels.
PATHOLOGICAL DIAGNOSIS
Binswanger's disease.
ACKNOWLEDGMENTS
This work was supported by National Institute on Aging Grant P50 AG16570 through the UCLA Alzheimer Disease Research Center.

FIGURE 1. T1-weighted MRI showing extensive white matter hyperintensities with compensatory ventricular enlargement

FIGURE 2. T2-weighted images showing similar white matter lesions

FIGURE 3. T1-weighted midsaggital MRI illustrating associated atrophy of the corpus callosum

FIGURE 4. Coronal section of fixed brain, parietal regionNote pronounced collapse and a “moth-eaten” appearance to the subcortical white matter. Overlying cortical ribbon appears focally thinned.

FIGURE 5. Section stained for myelin (Klüver-Barrera technique)Panel A shows relatively well preserved myelin bounded by cortical ribbon (at top and bottom of micrograph). By contrast, panel B shows a relatively pale, markedly demyelinated, and gliotic region of subcortical white matter. (Both panels, magnification×25).

FIGURE 6. Typical arteriosclerotic change in a white matter artery (arrow) seen in a region of demyelinationScattered lymphocytes are present in the vascular adventitia. (Hematoxylin and eosin stain; magnification×130).

FIGURE 7. Immunohistochemical features of brain parenchymaA: Glial fibrillary acidic protein (GFAP) immunostained section of a representative cerebral fragment. Cortex is at upper right, white matter at lower left. Both regions show dense collections of GFAP-immunoreactive astrocytes. B: CD68 immunostained section shows macrophages clustered around adventitia of a sclerotic artery (arrow), less prominent macrophages around other capillary size vessels, and immunoreactive microglia in rest of the white matter. C: Section immunostained with anti-CD3 (a T-cell marker) shows scattered immunoreactive cells in the white matter parenchyma, but several prominently labeled cells in an arterial wall (arrow). (Magnifications: A×25, B×65, C×130).
 |
 |
1 Hsich G, Kenney K, Gibbs CJ, et al: The 14-3-3 brain protein in cerebrospinal fluid as a marker for transmissible spongiform encephalopathies. N Engl J Med 1996; 335:924-930Crossref, Medline, Google Scholar
2 Mega MS, Cohenour RC: Akinetic mutism: disconnection of frontal-subcortical circuits. Neuropsychiatry Neuropsychol Behav Neurol 1997; 10:254-259Medline, Google Scholar
3 Collins S, Boyd A, Fletcher A, et al: Recent advances in the pre-mortem diagnosis of Creutzfeldt-Jakob disease. J Clin Neurosci 2000; 7:195-202Crossref, Medline, Google Scholar
4 Murakami N: Parkinsonism-dementia complex on Guam: overview of clinical aspects. J Neurol 1999; 246(suppl 2):1116-1118Google Scholar
5 Weinshenker B, Lucchinetti C: Acute leucoencephalopathies: differential diagnosis and investigation. The Neurologist 1998; 4:148-166Crossref, Google Scholar
6 Hachinski VC, Potter P, Merskey H: Leuko-araiosis. Arch Neurol 1987; 44:21-23Crossref, Medline, Google Scholar
7 Kidwell CS, Saver JL: Progress in understanding white matter hyperintensities. Stroke 1996; 27:1274-1282, 2338Crossref, Medline, Google Scholar
8 Breteler MMB, Van Swieten JC, Bots ML, et al: Cerebral white matter lesions, vascular risk factors, and cognitive function in a population-based study: the Rotterdam Study. Neurology 1994; 44:1246-1252Crossref, Medline, Google Scholar
9 Hunt AL, Orrison WW, Yeo RA, et al: Clinical significance of MRI white matter lesions in the elderly. Neurology 1989; 39:1470-1474Crossref, Medline, Google Scholar
10 Van Gijn J: Leukoaraiosis and vascular dementia. Neurology 1998; 51(3 suppl 3):S3-S8Google Scholar
11 Meguro K, Hatazawa J, Yamaguchi T, et al: Cerebral circulation and oxygen metabolism associated with subclinical periventricular hyperintensity as shown by magnetic resonance imaging. Ann Neurol 1990; 28:378-383Crossref, Medline, Google Scholar
12 Tohgi H, Yonezawa H, Takahashi S, et al: Cerebral blood flow and oxygen metabolism in senile dementia of Alzheimer's type and vascular dementia with deep white matter changes. Neuroradiology 1998; 40:131-137Crossref, Medline, Google Scholar
13 Janota I, Mirsen TR, Hachinski VC, et al: Neuropathologic correlates of leuko-araiosis. Arch Neurol 1989; 46:1124-1128Crossref, Medline, Google Scholar
14 Pullicino P, Benedict RHB, Capruso DX, et al: Neuroimaging criteria for vascular dementia. Arch Neurol 1996; 53:723-728Crossref, Medline, Google Scholar
15 Erkinjuntti T: Vascular dementia: challenge of clinical diagnosis. Int Psychogeriatrics 1997; 9:51-58Crossref, Medline, Google Scholar
16 Bondareff W, Raval J, Woo B, et al: Magnetic resonance imaging and the severity of dementia in older adults. Arch Gen Psychiatry 1990; 47:47-51Crossref, Medline, Google Scholar
17 Kertesz A, Polk M, Carr T: Cognition and white matter changes on magnetic resonance imaging in dementia. Arch Neurol 1990; 47:387-391Crossref, Medline, Google Scholar
18 Groves WC, Brandt J, Steinberg M, et al: Vascular dementia and Alzheimer's disease: is there a difference? A comparison of symptoms by disease duration. J Neuropsychiatry Clin Neurosci 2000; 12:305-315Link, Google Scholar
19 Liu CK, Miller BL, Cummings JL, et al: A quantitative MRI study of vascular dementia. Neurology 1992; 42:138-143Crossref, Medline, Google Scholar
20 Steingart A, Hachinski V, Lau C, et al: Cognitive and neurologic findings in demented patients with diffuse white matter lucencies on computed tomographic scan (leuko-araiosis). Arch Neurol 1987; 44:36-39Crossref, Medline, Google Scholar
21 Erkinjuntti T, Haltia M, Palo J, et al: Accuracy of the clinical diagnosis of vascular dementia: a prospective clinical and post-mortem neuropathological study. J Neurol Neurosurg Psychiatry 1988; 51:1037-1044Crossref, Medline, Google Scholar
22 Boone KB, Miller BI, Lesser IM, et al: Neuropsychological correlates of white-matter lesions in healthy elderly subjects: a threshold effect. Arch Neurol 1992; 49:549-554Crossref, Medline, Google Scholar
23 Yamauchi H, Fukuyama H, Shio H: Corpus callosum atrophy in patients with leukoaraiosis may indicate global cognitive impairment. Stroke 2000; 31:1515-1520Crossref, Medline, Google Scholar
24 Filley CM: The behavioral neurology of cerebral white matter. Neurology 1998; 50:1535-1540Crossref, Medline, Google Scholar
25 Junque C, Pujol J, Vendrell P, et al: Leuko-araiosis on magnetic resonance imaging and speed of mental processing. Arch Neurol 1990; 47:151-156Crossref, Medline, Google Scholar
26 Mendez MF, Cherrier MM, Perryman KM: Differences between Alzheimer's disease and vascular dementia on information processing measures. Brain Cogn 1997; 34:301-310Crossref, Medline, Google Scholar
27 Cummings JL: Frontal-subcortical circuits and human behavior. Arch Neurol 1993; 50:873-880Crossref, Medline, Google Scholar
28 Dichgans M, Mayer M, Uttner I, et al: The phenotype spectrum of CADASIL: clinical findings in 102 cases. Neurology 1998; 44:731-739Google Scholar
29 Mendez MF, Stanley TM, Medel NM, et al: The vascular dementia of Fabry's disease. Dementia Geriatr Cogn Disord 1997; 8:252-257Crossref, Medline, Google Scholar
30 Bornebroek M, Haan J, Van Buchem M, et al: White matter lesions and cognitive deterioration in presymptomatic carriers of the amyloid precursor protein gene codon 693 mutation. Arch Neurol 1996; 53:43-48Crossref, Medline, Google Scholar
31 Jen J, Cohen AH, Yue Q, et al: Hereditary endotheliopathy with retinopathy, nephropathy, and stroke (HERNS). Neurology 1997; 49:1322-1330Crossref, Medline, Google Scholar
32 Wright RA, Kokmen EA: Gradually progressive dementia without discrete cerebro-vascular events in a patient with Sneddon's syndrome. Mayo Clin Proc 1999; 74:57-61Crossref, Medline, Google Scholar
33 Kinkel WR, Jacobs L, Polachini I, et al: Subcortical arteriosclerotic encephalopathy (Binswanger's disease). Arch Neurol 1985; 42:951-959Crossref, Medline, Google Scholar
34 Summergrad P, Peterson B: Binswanger's disease (part I): the clinical recognition of subcortical arteriosclerotic encephalopathy in elderly neuropsychiatric patients. J Geriatr Psychiatry Neurol 1989; 2:123-133Crossref, Medline, Google Scholar
35 Hurley RA, Hidekazu T, Akiguchi I, et al: Binswanger's disease: an ongoing controversy. J Neuropsychiatry Clin Neurosci 2000; 12:301-304Link, Google Scholar
36 Román GC: From UBOs to Binswanger's disease: impact of magnetic resonance imaging on vascular dementia research. Stroke 1966; 27:1269-1273Google Scholar
37 Hanyu H, Asano T, Sakurai H, et al: Magnetization transfer ratio in cerebral white matter lesions of Binswanger's disease. J Neurol Sci 1999; 166:85-90Crossref, Medline, Google Scholar
38 Choi SH, Na DL, Chung CS, et al: Diffusion-weighted MRI in vascular dementia. Neurology 2000; 54:83-89Crossref, Medline, Google Scholar
39 Puls I, Hauck K, Demuth K, et al: Diagnostic impact of cerebral transit time in the identification of microangiopathy in dementia: a transcranial ultrasound study. Stroke 1999; 30:2291-2295Crossref, Medline, Google Scholar
40 Matteis M, Silvestrini M, Troisi E, et al: Cerebral hemodynamic patterns during stimuli tasks in multi-infarct and Alzheimer types of dementia. Acta Neurol Scand 1998; 97:374-378Medline, Google Scholar
41 Akiguchi I, Tomimoto H, Suenaga T, et al: Alterations in glia and axons in the brains of Binswanger's disease patients. Stroke 1997; 28:1423-1429Crossref, Medline, Google Scholar
42 Fisher CM: Binswanger's encephalopathy: a review. J Neurol 1989; 236:65-79Crossref, Medline, Google Scholar
43 Dubas F, Gray F, Roullet E, et al: Leucoencephalopathies arteriopathique (17 cas anatomo-cliniques). Rev Neurol 1985; 141:93-108Medline, Google Scholar
44 Babikian V, Ropper AH: Binswanger's disease: a review. Stroke 1987; 18:2-12Crossref, Medline, Google Scholar

