
Influence of the Apolipoprotein E Type 4 Allele on Cerebral Glucose Metabolism in Alzheimer's Disease Patients
Abstract
It is still unclear whether apolipoprotein E epsilon 4 (APOE E4) influences the cerebral glucose metabolism abnormalities found in Alzheimer's disease (AD), although APOE E4 is a well-known risk factor for AD. [18F]Fluorodeoxyglucose PET was conducted in patients with very mild (n=17), mild (n=27), and moderate-to-severe (n=19) AD. The presence of APOE E4 was associated with greater reduction of glucose metabolism in the left inferior temporal region in very mild AD but neither in mild nor in moderate-to-severe AD. These findings favor the hypothesis that APOE E4 is related mainly to the development of AD, not to its progression.
The apolipoprotein E epsilon 4 allele (APOE E4) is a well-known risk factor for late-onset familial Alzheimer's disease (AD)1 as well as for sporadic AD,2–4 and the inheritance of the APOE E4 allele is believed to lower the age at onset in a dose-dependent manner.5 Previous studies have strongly suggested that APOE E4 might play an important role in the pathogenesis of AD. APOE E4 may be associated with a faster deposition of the amyloid β protein,6 and isoforms-specific interactions of APOE with the tau protein7 and cholinergic function8 may exist. Furthermore, several lines of clinical studies have reported that more severe cognitive,9,10 gross structural,11,12 and electrophysiological13,14 abnormalities were associated with the presence of APOE E4.
By use of [18F]fluorodeoxyglucose positron emission tomography (PET), characteristic abnormalities of the cerebral metabolic rate of glucose metabolism (CMRglc) in the temporal, parietal, cingulate, and sometimes frontal regions have been observed in patients with AD15,16 as well as in persons who are at risk for AD.17 In studies concerning the influence of APOE E4 on CMRglc, APOE E4 carriers showed a more pronounced reduction of CMRglc in the temporal, parietal, and prefrontal regions in normal elderly subjects with a higher risk of AD.18,19 However, later PET studies have repeatedly failed to demonstrate the association of APOE E4 allele with further reduction of CMRglc in patients with AD.20,21
Although the pathogenic role of APOE E4 in AD has been readily recognized, there is still controversy as to whether APOE E4 affects the progression of AD. Some prospective studies have reported that there was no difference in the rate of cognitive decline in relation to the inheritance of APOE E4.22,23 These results suggest that APOE E4 might be involved in the development of AD but does not influence the course of AD once the disease has developed. Therefore, it is possible to hypothesize that the effect of APOE E4 on CMRglc in AD might vary at different stages and that the change of CMRglc associated with the inheritance of APOE E4 is prominent only in the early stage of AD, disappearing with the progression of the disease. We suspected that the subjects with AD in previous PET studies were heterogeneous in clinical severity; however, no clear description was given.20,21
In this study, therefore, we tried to observe the effect of APOE E4 on CMRglc in patients with AD after classifying the subjects into clinically defined different severity groups. The objective of this study was to demonstrate whether the effect of APOE E4 on CMRglc is present in subjects with early-stage AD and disappears in those at later stages of the disease.
METHODS
Subjects
All of the recruited patients met both National Institute of Neurological and Communicative Disorders and Stroke–Alzheimer's Disease and Related Disorders Association (NINCDS-ADRDA) criteria for probable AD24 and DSM-IV criteria for dementia of the Alzheimer's type.25 Clinical evaluations were made by using the Korean version of the Consortium to Establish a Registry for Alzheimer's Disease Assessment Packet.26 Patients underwent psychiatric, general physical, and neurological examinations; standard neuropsychological tests including the Mini-Mental State Examination (MMSE);27 routine laboratory tests; and MRI of the brain to exclude other causes of dementia. [18F]fluorodeoxyglucose PET was performed in each subject. Patients who scored more than 4 on the Modified Ischemic Scale28 were excluded from the study, as were patients with left or both handedness. The severity of the AD was assessed by using the Clinical Dementia Rating Scale (CDRS),29 and the subjects were divided into three different clinical severity groups of very mild (CDRS=0.5, n=17), mild (CDRS=1, n=27), and moderate to severe (CDRS=2 or 3, n=19). The APOE E4+ group consisted of subjects with one or more APOE E4 alleles, and the APOE e4– group included the subjects without any APOE E4 allele. All subjects provided their informed consent for participation in the study.
Determination of the Apolipoprotein E Genotype
Genomic DNA was extracted from venous blood, and APOE genotyping was done according to the method described by Wenham et al.30 The sequences of the primer pair were 5′-TCCAAGGAGCTGCAGGCGCGCCA-3′ as the upstream primer and 5′-ACAGAATTCGCCCCGGCCTGGTACACTGCCA-3′ as the downstream primer. An initial denaturation at 94° C for 10 min was followed by 30 cycles of annealing at 65° C for 0.5 min, extension at 72° C for 2 min, denaturation at 94° C for 1 min, and final extension at 72° C for 5 min. The APOE genotypes were identified by the detection of a unique combination of fragment sizes: 91-bp and 81-bp fragments for E2, 91-bp and 48-bp fragments for E3, and 72-bp and 48-bp fragments for E4.
[18F]Fluorodeoxyglucose PET
PET studies were performed by using the ECAT EXACT 47 scanner (Siemens-CTI, Knoxville, TN, USA), which has an intrinsic resolution of 5.2 mm full width at half maximum (FWHM) and images 47 contiguous planes simultaneously with a thickness of 3.4 mm, for a longitudinal field of view equaling 16.2 cm. Before [18F]fluorodeoxyglucose administration, transmission scanning was performed, using three germanium-68 rod sources for attenuation correction. Static emission scans were begun 30 minutes after the injection of 370 MBq (10 mCi) [18F]fluorodeoxyglucose and were continued for 30 minutes. Transaxial images were reconstructed by means of a filtered back-projection algorithm employing a Shepp-Logan filter with a cutoff frequency of 0.3 cycles/pixel as 128×128×47 matrices with a size of 2.1×2.1×3.4 mm.
Image Analysis
Image data were analyzed by using SPM96 (Statistical Parametric Mapping 96; Wellcome Department of Cognitive Neurology, London, UK) implemented in Matlab (Mathworks Inc., Sherborn, MA, USA). Before statistical analysis, all of the images were spatially normalized into the MNI (Montreal Neurological Institute, McGill University, Montreal, Canada) standard template to correct for intersubject anatomical variability. Affine transformation was performed to determine the 12 optimal parameters necessary to register the brain on the template. Subtle differences between the transformed image and the template were removed by the nonlinear registration method, employing the weighted sum of the predefined smooth basis functions used in discrete cosine transformation. Spatially normalized images were smoothed by convolution with an isotropic Gaussian kernel with 16 mm FWHM. The count of each voxel was normalized to the pontine activity, under the assumption that energy metabolism in the pons is minimally affected in Alzheimer's disease.31 After spatial and count normalization, the significant difference in cerebral glucose metabolism between the APOE E4+ and APOE e4– groups was estimated at every voxel on the basis of a general linear model using appropriate contrasts. The effects of age were accounted for by using analysis of covariance (ANCOVA) with age as a covariate at each voxel. Using the means and variances that were adjusted with ANCOVA for both groups, t-tests were performed at every voxel and transformed to the standard Gaussian distribution (Z score). The voxels with uncorrected P-values of less than 0.005 (i.e., Z>2.58) were considered to show statistically significant differences.
RESULTS
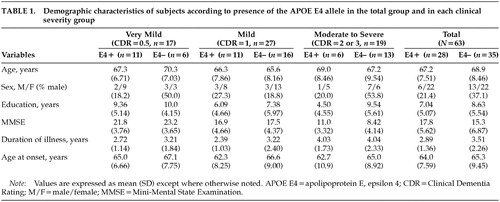
A total of 63 patients with AD participated in this study. Twenty-eight of them were APOE E4 carriers and 35 were noncarriers. There were no significant differences on two-tailed t-tests (means for age, education, MMSE-K score, duration of illness, and age at onset) or chi-square tests (sex ratio) between the APOE E4+ and APOE e4– groups in total, or in any of the clinical severity groups (Table 1). Although there was a tendency to lower age at onset in the APOE E4+ groups, the differences were not statistically significant.
In comparing CMRglc between the APOE E4+ group and the APOE e4– group among the total number of AD patients, there was no voxel with a significant difference in glucose metabolism on SPM analysis. However, when the analyses were performed in each clinical severity group, regions where the APOE E4+ group showed greater decline of CMRglc than the APOE e4– group were observed. In the very mild clinical severity group, SPM showed a significantly decreased CMRglc in the left inferior temporal regions (3,247 voxels, P<0.005, uncorrected for multiple comparison) in the APOE E4+ group compared with the APOE e4– group. The left inferior temporal regions included three peaks of maximal decline of CMRglc, and each peak corresponded to Brodmann areas 20, 37, 37 (P=0.0002, P=0.0008, P=0.0013, respectively). The SPM results and coordinates of maximal decline are shown in Table 2 and Figure 1. There was no brain region in which the APOE e4– group showed greater decline of CMRglc than the APOE E4+ group in the patients with very mild clinical severity. In the mild group as well as the moderate-to-severe group, there were no regional differences in CMRglc between the APOE E4+ and APOE e4– groups.
DISCUSSION
The association of APOE E4 with greater deficits of CMRglc has been consistently reported in normal elderly subjects at high risk for AD.18,19 However, previous studies attempting to elucidate the effect of APOE E4 on CMRglc in patients with AD have failed to do so.20,21 Corder et al.,21 studying the rCMRglc in 31 patients with AD, reported no association of APOE E4 allele with specific deficits in brain metabolism. The clinical severity of their study population was heterogeneous, in that 12 of the 31 subjects had only mild memory complaints. In a study of 83 AD patients by Hirono et al.,20 the clinical severity was not discussed in great detail. These results, in fact, are consistent with our results in that no difference in CMRglc between the APOE E4+ and APOE e4– groups among the total number of AD patients was observed. In our study, 17 of the 63 subjects had very mild dementia. Therefore, the breakdown of our subjects was similar to that in the Corder et al.21 study.
When further comparisons of CMRglc between the APOE E4+ and APOE e4– groups were conducted after classifying the subjects into different clinical severity groups, the APOE E4+ group showed greater reduction of CMRglc in the left inferior temporal region, but only in the subjects with very mild clinical severity. No difference in CMRglc was observed in the mild and moderate-to-severe groups. The subjects with very mild dementia described in our study clearly met the NINCDS-ADRDA and DSM-IV criteria for probable AD. Therefore, they differed from the subjects described in the Small et al. study,19 who were normal elderly people at risk for AD who met the diagnostic criteria for age-associated memory impairment, but not for AD. Our results provided direct evidence that the APOE E4 allele influences the dysfunction of the brain in AD.
Although it is readily recognized that APOE E4 lowers the age at onset and increases the risk of AD, there is still controversy as to whether APOE E4 influences the rate of progression of AD. Several studies have reported that the APOE genotype did not influence the rate of cognitive decline in AD.22,23 However, another study reported accelerated cognitive decline in APOE E4 homozygotes with AD.32 Still other studies have reported even better prognoses in APOE E4 carriers.33,34 The results of our study favor the hypothesis that APOE E4 is not associated with the progression of AD, but is related only to the developmental stage of the disease.
The finding of APOE e4– related hypometabolism in the left temporal region corresponds with clinical, magnetic resonance–based imaging, and neuropathologic studies. Several studies have shown that the presence of APOE E4 in AD was related to more severe impairment of the types of memory that are the function of the hippocampus and temporal lobe structures, such as delayed recall.9,10 There have been at least two MRI studies in which a larger volume loss of the medial temporal lobe structures was seen in patients with AD homozygous for APOE E4.11,12 In a neuropathologic study, an increased deposition of the amyloid β peptide35 and decreased choline acetyltransferase activity36 of the temporal lobe were reported to be associated with the presence of the APOE E4 allele.
The methodological considerations in this study include the statistical issues in analyzing the PET data by using SPM96. Although a successful clinical validation study using SPM was reported recently,37 the danger of increased alpha error due to multiple comparisons still exists. Because a standard guideline for proper acquisition of statistical inference has not yet been established, we applied a commonly used setting (P<0.005, SPM [Z]>2.58) for explorative purposes. However, the SPM (Z) score of the observed cluster (left inferior temporal region) was actually more than 3.09 (P<0.001). Therefore, it is less likely that significant clusters were observed by chance.
In this study, we showed that the presence of the APOE E4 allele was associated with greater reduction of cerebral glucose metabolism in patients with AD. Furthermore, reduction of glucose metabolism was prominent only in the very early stage of AD; the effect of APOE E4 on CMRglc disappeared in the more advanced stages of the disease. These results confirm that the deficit of CMRglc in AD is influenced by APOE E4 and favor the hypothesis that APOE E4 is mainly related to the development of AD, but not to its progression. Further longitudinal studies are necessary to determine the effects of APOE E4 on disease progression of AD.
ACKNOWLEDGMENTS
This study received support from the Seoul National University Hospital (Grant 04-98-045) and Biotech 2000 (Grants 97-N1-02-03-A-12 and 98-N1-02-03-A-12), supported by the Ministry of Science and Technology of the Korean Government.

FIGURE 1. Three-dimensional surface projection maps of reduced cerebral glucose metabolism (indicated in yellow) in 11 patients with very mild AD with the APOE E4 allele compared with that in 6 similar patients without the APOE E4 allele, superimposed on spatially standardized and volume-rendered MRIs of the brain. Analysis of covariance was performed using SPM96 with age as a covariate (P<0.005, uncorrected for multiple comparisons).
 |
 |
1 Strittmatter WJ, Saunders AM, Schmechel D, et al: Apolipoprotein E: high-avidity binding to β-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer's disease. Proc Natl Acad Sci USA 1993; 90:1977-1981Crossref, Medline, Google Scholar
2 Rebeck GW, Reiter JS, Strickland DK, et al: Apolipoprotein E in sporadic Alzheimer's disease: allelic variation and receptor interactions. Neuron 1993; 11:575-580Crossref, Medline, Google Scholar
3 Saunders AM, Strittmatter WJ, Schmechel D, et al: Association of apolipoprotein E allele ε4 with late-onset familial and sporadic Alzheimer's disease. Neurology 1993; 43:1467-1472Crossref, Medline, Google Scholar
4 Ueki A, Kawano M, Namba Y, et al: A high frequency of apolipoprotein E4 isoprotein in Japanese patients with late-onset non-familial Alzheimer's disease. Neurosci Lett 1993; 163:166-168Crossref, Medline, Google Scholar
5 Blacker D, Haines JL, Rodes L, et al: ApoE-4 and age at onset of Alzheimer's disease: the NIMH Genetics Initiative. Neurology 1997; 48:139-147Crossref, Medline, Google Scholar
6 Schmechel DE, Saunders AM, Strittmatter WJ, et al: Increased amyloid β-peptide deposition in cerebral cortex as a consequence of an apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci USA 1993; 90:9649-9653Crossref, Medline, Google Scholar
7 Strittmatter, WJ, Saunders AM, Goedert M, et al: Isoform-specific interactions of apolipoprotein E with microtubule-associated protein tau: implications for Alzheimer disease. Proc Natl Acad Sci USA 1994; 91:11183-11186Crossref, Medline, Google Scholar
8 Poirier J, Delisle M-C, Quirion R, et al: Apolipoprotein ε4 allele as a predictor of cholinergic deficits and treatment outcome in Alzheimer's disease. Proc Natl Acad Sci USA 1995; 92:12260-12264Crossref, Medline, Google Scholar
9 Smith GE, Bohac DL, Waring SC, et al: Apolipoprotein E genotype influences cognitive “phenotype” in patients with Alzheimer's disease but not in healthy control subjects. Neurology 1998; 50:355-362Crossref, Medline, Google Scholar
10 Lehtovirta M, Soininen H, Helisalmi S, et al: Clinical and neuropsychological characteristics in familial and sporadic Alzheimer's disease: relation to apolipoprotein E polymorphism. Neurology 1996; 46:413-419Crossref, Medline, Google Scholar
11 Juottonen K, Lehtovirta M, Helisalmi S, et al: Major decrease in the volume of the entorhinal cortex in patients with Alzheimer's disease carrying the apolipoprotein E epsilon4 allele. J Neurol Neurosurg Psychiatry 1998; 65:322-327Crossref, Medline, Google Scholar
12 Lehtovirta M, Laakso MP, Soininen H, et al: Volumes of hippocampus, amygdala and frontal lobe in Alzheimer patients with different apolipoprotein E genotypes. Neuroscience 1995; 67:65-72Crossref, Medline, Google Scholar
13 Jelic V, Julin P, Shigeta M, et al: Apolipoprotein E epsilon4 allele decreases functional connectivity in Alzheimer's disease as measured by EEG coherence. J Neurol Neurosurg Psychiatry 1997; 63:59-65Crossref, Medline, Google Scholar
14 Lehtovirta M, Partanen J, Kononen M, et al: Spectral analysis of EEG in Alzheimer's disease: relation to apolipoprotein E polymorphism. Neurobiol Aging 1996; 17:523-526Crossref, Medline, Google Scholar
15 Duara R, Grady C, Haxby J, et al: Positron emission tomography in Alzheimer's disease. Neurology 1986; 36:879-887Crossref, Medline, Google Scholar
16 Foster NL, Chase TN, Fedio P, et al: Alzheimer's disease: focal changes shown by positron emission tomography. Neurology 1983; 33:961-965Crossref, Medline, Google Scholar
17 Pietrini P, Azari NP, Grady CL, et al: Pattern of cerebral metabolic interactions in a subject with isolated amnesia at risk for Alzheimer's disease: a longitudinal evaluation. Dementia 1993; 4:94-101Medline, Google Scholar
18 Reiman EM, Caselli RJ, Yun LS, et al: Preclinical evidence of Alzheimer's disease in persons homozygous for the ε4 allele for apolipoprotein E. N Engl J Med 1996; 334:752-758Crossref, Medline, Google Scholar
19 Small GW, Mazziotta JC, Collins MT, et al: The effect of apolipoprotein E type 4 allele on cerebral glucose metabolism in relatives at risk for Alzheimer disease. JAMA 1995; 273:942-947Crossref, Medline, Google Scholar
20 Hirono N, Mori E, Yasuda M, et al: Lack of association of apolipoprotein E epsilon4 allele dose with cerebral glucose metabolism in Alzheimer's disease. Alzheimer Dis Assoc Disord 1998; 12:362-367Crossref, Medline, Google Scholar
21 Corder EH, Jelic V, Basun H, et al: No difference in cerebral glucose metabolism in patients with Alzheimer disease and differing apolipoprotein E genotypes. Arch Neurol 1997; 54:273-277Crossref, Medline, Google Scholar
22 Murphy GM, Taylor J, Kraemer HC, et al: No association between apolipoprotein E ε4 allele and rate of decline in Alzheimer's disease. Am J Psychiatry 1997; 154:603-608Crossref, Medline, Google Scholar
23 Dal Forno G, Rasmusson DX, Brandt J, et al: Apolipoprotein E genotype and rate of decline in probable Alzheimer's disease. Arch Neurol 1996; 53:345-350Crossref, Medline, Google Scholar
24 Mckhann G, Drachman D, Folstein M, et al: Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA work group under the auspices of Department of Health and Human Services Task Force on Alzheimer's disease. Neurology 1984; 11:939-944Crossref, Google Scholar
25 American Psychiatric Association: Diagnostic and Statistical Manual of Mental Disorders, 4th ed. Washington, DC, American Psychiatric Association, 1994Google Scholar
26 Lee JH, Lee KU, Lee DY, et al: Development of the Korean version of the Consortium to Establish a Registry for Alzheimer's Disease Assessment Packet (CERAD-K): clinical and Neuropsychological Assessment Batteries. J Gerontol B Psychol Sci 2002; 57:47-53Crossref, Google Scholar
27 Folstein MF, Folstein SE, McHugh PR: “Mini-Mental State”: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975; 12:189-198Crossref, Medline, Google Scholar
28 Rosen WG, Terry RD, Fuld PA, et al: Pathological verification of ischemic score in differentiation of dementias. Ann Neurol 1980; 7:486-488Crossref, Medline, Google Scholar
29 Hughes CP, Berg L, Danziger W, et al: A new clinical scale for the staging of dementia. Br J Psychiatry 1982; 140:566-572Crossref, Medline, Google Scholar
30 Wenham PR, Price WH, Blandell G: Apolipoprotein E genotyping by one-stage PCR. Lancet 1991; 337:1158-1159Crossref, Medline, Google Scholar
31 Minoshima S, Frey KA, Foster NL, et al: Preserved pontine glucose metabolism in Alzheimer disease: a reference region for functional brain image (PET) analysis. J Comput Assist Tomogr 1995; 19:541-547Crossref, Medline, Google Scholar
32 Craft S, Teri L, Edland SD, et al: Accelerated decline in apolipoprotein E-epsilon4 homozygotes with Alzheimer's disease. Neurology 1998; 51:149-153Crossref, Medline, Google Scholar
33 Basun H, Grut M, Winblad B, et al: Apolipoprotein epsilon 4 allele and disease progression in patients with late-onset Alzheimer's disease. Neurosci Lett 1995; 183:32-34Crossref, Medline, Google Scholar
34 Stern Y, Brandt J, Albert M, et al: The absence of an apolipoprotein ε4 allele is associated with a more aggressive form of Alzheimer's disease. Ann Neurol 1997; 41:615-620Crossref, Medline, Google Scholar
35 McNamara MJ, Gomez-Isla T, Hyman BT: Apolipoprotein E genotype and deposits of Abeta40 and Abeta42 in Alzheimer's disease. Arch Neurol 1998; 55:1001-1004Crossref, Medline, Google Scholar
36 Beffert U and Poirier J: Apolipoprotein E, plaques, tangles and cholinergic dysfunction in Alzheimer's disease. Ann NY Acad Sci 1996; 777:166-174Crossref, Medline, Google Scholar
37 Signorini M, Paulesu E, Friston K, et al: Rapid assessment of regional cerebral metabolic abnormalities in single subjects with quantitative and nonquantitative [18F] FDG PET: a clinical validation of statistical parametric mapping. Neuroimage 1999; 9:63-80Crossref, Medline, Google Scholar
38 Talairach J, Tournoux P. Co-planar Stereotaxic Atlas of the Human Brain. New York, Thieme Medical, 1988Google Scholar

