
Cortical Inhibition in Alcohol Dependence
Alcohol dependence as defined by DSM-IV criteria is a psychiatric disorder that affects approximately 13% of the population at some point in life. This prevalence increases to 20% if individuals who have never consumed 12 or more drinks in any one year of life are excluded.1 Approximately half of all homicides and motor vehicle–related deaths involve alcohol, as do one-fourth of all suicides. Comorbid psychiatric conditions are common. The estimated social impact of alcohol dependence is $100 billion in health care costs, lost wages, and family disruption.2 To understand this enormous public health concern, researchers are studying alcoholism from the sociological to the molecular level. (A PubMed search on “alcohol” captures more than 68,000 references in the past five years alone.) A few research areas include the neurobiology of craving and tolerance, changes in cortical neurochemistry with dependence and withdrawal, and the development of medications to reverse intoxication and to prevent craving.
A genetic predisposition or vulnerability to development of alcohol dependence has been clearly demonstrated. There is a 3- to 9-fold increased risk in first-degree relatives of alcohol-dependent patients compared with the general population.3 The genetic vulnerability to alcohol dependence is 0.50 to 0.60. Two chromosomal linkage studies have found strong evidence for possible candidate genes for alcohol dependence on chromosome 4 and lesser evidence for chromosomes 11, 1, 7, and 2.4
It has been recognized since the case of Phineas Gage was understood that the orbitofrontal cortex is a critical part of a circuit responsible for inhibition and for regulation of social behavior. It has been suggested that there is a relationship between alcohol dependence and the orbitofrontal cortex. This theory proposes a shift in the general excitability of the brain, perhaps as a result of decreased inhibition (disinhibition). Alcohol-dependent persons score high on several measures likely to reflect disinhibition, including exploratory excitability, impulsiveness, extravagance, and disorderliness.5 In addition, there is evidence of abnormal brain processing in both persons who are alcohol dependent and individuals at high risk for alcoholism.
Evidence for abnormal processing has been obtained by using event-related potentials (ERPs). ERPs are the summed electrical activity recorded from scalp electrodes after a stimulus presentation. A variety of situations or tasks are used in this type of study, and the ERP has a characteristic shape for each. A very simple task commonly used requires monitoring a series of frequent visual shapes (e.g., squares) and responding by pushing a button whenever a designated different shape (e.g., a triangle) is presented (“oddball” task). The different, rarely occurring shape is the target. In normal individuals the positive waveform that occurs approximately 300 ms after the stimulus onset (P300 waveform) is much larger following presentation of a target compared with a nontarget in this task. In alcohol-dependent subjects, the P300 waveform to presentation of targets is much smaller than in normal individuals.6 Importantly, it is reduced more in those with alcohol dependence who are from high-risk families than in those with no family history of alcohol abuse. It is also abnormal in individuals from high-risk families who are alcohol naive.7 This waveform is thought to reflect inhibitory processes in cortex. Thus, the decreased size in patients with alcohol dependence and persons at risk may indicate a global or regional lack of cortical inhibition. The diminished increase when a target is presented may indicate deficits in processing of information.8
Electrophysiological studies of more complex tasks also support this view. CNS inhibitory state can be assessed experimentally by use of a cued continuous performance test in which a motor response is required to one situation (Go condition) but must be suppressed to others (No-Go condition). The ERP to each condition in this task is influenced by the complexity of the Go/No-Go decision.9 In a simple version of this task a series of uppercase and lowercase letters is presented (usually all the same letter). One is designated the Go and the other the No-Go condition. In normal individuals the P300 waveform to the Go condition is larger than to the No-Go condition for this task, similar to results of the “oddball” task described previously. In contrast, subjects who are alcohol dependent do not show any increase in the P300 waveform to the Go condition, and they have lower P300 amplitudes overall.9
In a more difficult Go/No-Go task, a series of letters is presented in which one letter is designated as the primer. When the target letter follows the primer, the subject responds by pushing a button (Go condition). When any other letter follows the primer, the subject must inhibit responding (No-Go condition).10 The P300 component of the ERP recorded following a stimulus in this task is localized to both frontal and parietal regions.11 The frontal activation is larger in the No-Go condition than in the Go condition. A reasonable interpretation of this finding is that the frontal activation reflects the inhibition that is required for response suppression during the No-Go condition.11 Persons who are alcohol dependent have less frontal activation during this task than normal individuals, an indication that frontal lobe control of response inhibition is reduced.10
Although alcohol interacts with many neurotransmitters in both excitatory and inhibitory pathways, its potentiation of benzodiazepines (BZDs) and interactions with γ-aminobutyric acid (GABA) provide key support for this theory. GABA is the major inhibitory neurotransmitter in the brain. Postmortem studies have found regional changes in BZD receptor density in alcohol-dependent persons.12 The BZD receptor is a site on the GABAA receptor. Recent in vivo SPECT and PET studies have found decreased BZD receptor density in frontal, anterior cingulate, parietal, temporal, and cerebellar cortices in alcohol-dependent subjects compared with control subjects.13,14 These differences were present even in alcohol-dependent subjects who had been abstinent for prolonged periods prior to the evaluation. A similar pattern of differences was found when abstinent alcohol-dependent subjects were compared with nondependent alcohol users—suggesting that the differences in BZD receptor density are intrinsic rather than a result of alcohol toxicity.13 A pilot study using magnetic resonance spectroscopy found that cortical GABA levels were lower in alcohol-dependent subjects than in control subjects. In contrast, cortical glutamate levels were similar.15 Because GABA is an inhibitory neurotransmitter, this decrease in GABAergic transmission (both receptors and absolute levels) might underlie the diminished inhibition or hyperexcitability that has been found in alcohol-dependent individuals and persons at high risk for substance abuse.
Parallel results have been found with PET: alcohol-dependent patients usually have decreased metabolism in frontal, temporal (left), and parietal cortices.16,17 As alcohol-dependent patients detoxify, cortical metabolism improves.17 The greatest changes were obtained between 4 and 8 weeks after alcohol withdrawal. Orbitofrontal and dorsolateral prefrontal cortices showed the most recovery, basal ganglia the least.
Alcohol-dependent patients also have an altered cerebral metabolic response to BZD administration.18 In normal subjects, BZD administration decreases both global and regional brain metabolism. Measures taken during the first month of detoxification found less than normal metabolic depression in response to lorazepam administration in the thalamus, basal ganglia, and orbitofrontal cortex in alcohol-dependent subjects.18 Diminished responses may still be present at 11 weeks.19 Interestingly, the early changes correlated with both cerebellar metabolism at baseline and BZD (and therefore GABAA) receptor density. The cerebellum has direct GABAergic projections to thalamus, basal ganglia, and orbitofrontal cortex. Thus, it is possible that abnormalities in cerebellar input to the orbitofrontal circuit (which includes all three regions) lead to disinhibition and/or compulsive behaviors in alcohol-dependent patients. It is of interest that the depression in cerebellar function (as measured by both metabolic rate and motor coordination) normally induced by lorazepam administration is also diminished in alcohol-naive subjects with positive family histories for alcohol abuse.20
Although these studies are limited to small groups of subjects and many were performed early in detoxification, they may cast light on a portion of what underlies this devastating illness. The involvement with GABA is, of course, only one aspect of the effect of alcohol on the brain. Alcohol has major interactions with many aspects of brain function, including most or all of the major neurotransmitters. As the biological causes and effects of alcohol dependence come to be understood more fully, sophisticated and successful treatments can be developed—just as the discovery of cross-tolerance of alcohol and BZDs has led to decreased mortality from alcohol withdrawal.
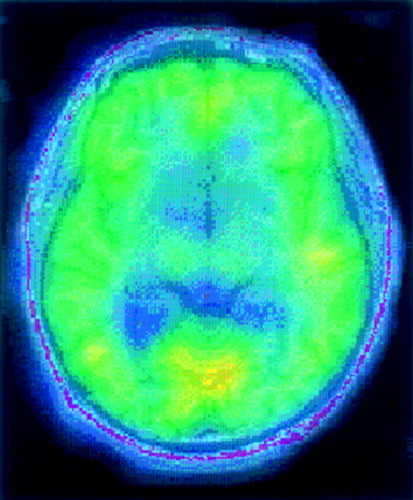
A Transaxial slice of a single-photon emission computed tomography (SPECT) study of benzodiazepine (BZD) receptor binding (iomazenil [123I]) superimposed on the corresponding MRI slice from a 56-year-old alcohol-dependent male who had been sober for 95 days. BZD binding in alcohol-dependent subjects is decreased in a number of brain regions compared with control subjects, as summarized below.
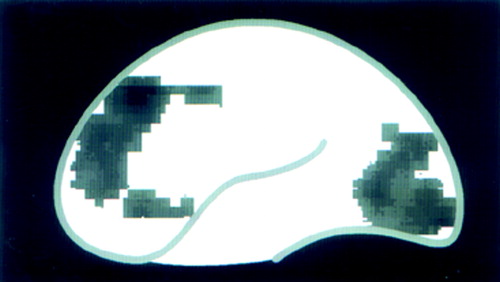
B Sagittal projection view (frontal cortex to the left, occipital cortex to the right) summarizes regions where BZD receptor distribution was lower in alcohol-dependent subjects compared with control subjects. Although BZD receptor binding was lower in all regions, it was significantly lower in prefrontal cortex, anterior cingulate cortex, and cerebellum.
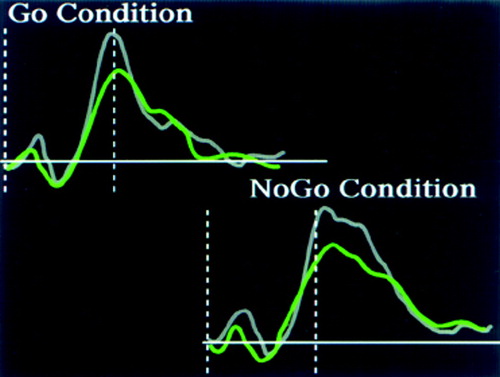
C Event-related potentials (ERPs) obtained by using a cued continuous performance test (Go/No-Go task). Note the decrease in the responses of the alcohol-dependent subjects (indicated in green) compared with control subjects (indicated in gray). The dashed lines indicate the time of target presentation and the time of peak response (300 ms, P300). The alcohol-dependent subjects had diminished frontal activation during both conditions, indicating diminished inhibition (disinhibition).
1 Grant BF: Prevalence and correlates of alcohol use and DSM-IV alcohol dependence in the United States: results of the National Longitudinal Alcohol Epidemiologic Survey. J Stud Alcohol 1997; 58:464–473Crossref, Medline, Google Scholar
2 Litten RZ, Allen JP: Advances in development of medications for alcoholism treatment. Psychopharmacol (Berl) 1998; 139:20–33Crossref, Medline, Google Scholar
3 McCaul ME: Substance abuse vulnerability in offspring of alcohol and drug abusers. NIDA Res Monogr 1998; 169:188–208Medline, Google Scholar
4 Enoch MA, Goldman D: Genetics of alcoholism and substance abuse. Psychiatr Clin North Am 1999; 22:289–299Crossref, Medline, Google Scholar
5 Wills TA, Vaccaro D, McNamara G: Novelty seeking, risk taking, and related constructs as predictors of adolescent substance use: an application of Cloninger's theory. J Subst Abuse 1994; 6:1–20Crossref, Medline, Google Scholar
6 Porjesz B, Begleiter H: Genetic basis of event-related potentials and their relationship to alcoholism and alcohol use. J Clin Neurophysiol 1998; 15:44–57Crossref, Medline, Google Scholar
7 Begleiter H, Porjesz B, Bihari B, et al: Event-related brain potentials in boys at risk for alcoholism. Science 1984; 225:1493–1496Google Scholar
8 Begleiter H, Porjesz B: What is inherited in the predisposition toward alcoholism? A proposed model. Alcohol Clin Exp Res 1999; 23:1125–1135Google Scholar
9 Cohen HL, Porjesz B, Begleiter H, et al: Neurophysiological correlates of response production and inhibition in alcoholics. Alcohol Clin Exp Res 1997; 21:1398–1406Google Scholar
10 Fallgatter AJ, Wiesbeck GA, Weijers HG, et al: Event-related correlates of response suppression as indicators of novelty seeking in alcoholics. Alcohol Alcohol 1998; 33:475–481Crossref, Medline, Google Scholar
11 Strik WK, Fallgatter AJ, Brandeis D, et al: Three-dimensional tomography of event-related potentials during response inhibition: evidence for phasic frontal lobe activation. Electroencephalogr Clin Neurophysiol 1998; 108:406–413Crossref, Medline, Google Scholar
12 Freund G, Ballinger WE: Loss of synaptic receptors can precede morphologic changes induced by alcoholism. Alcohol Alcohol Suppl 1991; 1:385–391Medline, Google Scholar
13 Lingford-Hughes AR, Acton PD, Gacinovic S, et al: Reduced levels of GABA-benzodiazepine receptor in alcohol dependency in the absence of grey matter atrophy. Br J Psychiatry 1998; 173:116–122Crossref, Medline, Google Scholar
14 Gilman S, Koeppe RA, Adams K, et al: Positron emission tomographic studies of cerebral benzodiazepine-receptor binding in chronic alcoholics. Ann Neurol 1996; 40:163–171Crossref, Medline, Google Scholar
15 Behar KL, Rothman DL, Petersen KF, et al: Preliminary evidence of low cortical GABA levels in localized 1H-MR spectra of alcohol-dependent and hepatic encephalopathy patients. Am J Psychiatry 1999; 156:952–954Crossref, Medline, Google Scholar
16 Volkow ND, Hitzemann R, Wang GJ, et al: Decreased brain metabolism in neurologically intact healthy alcoholics. Am J Psychiatry 1992; 149:1016–1022Google Scholar
17 Volkow ND, Wang GJ, Hitzemann R, et al: Recovery of brain glucose metabolism in detoxified alcoholics. Am J Psychiatry 1994; 151:178–183Crossref, Medline, Google Scholar
18 Volkow ND, Wang GJ, Hitzemann R, et al: Decreased cerebral response to inhibitory neurotransmission in alcoholics. Am J Psychiatry 1993; 150:417–422Crossref, Medline, Google Scholar
19 Volkow ND, Wang GJ, Overall JE, et al: Regional brain metabolic response to lorazepam in alcoholics during early and late alcohol detoxification. Alcohol Clin Exp Res 1997; 21:1278–1284Google Scholar
20 Volkow ND, Wang GJ, Begleiter H, et al: Regional brain metabolic response to lorazepam in subjects at risk for alcoholism. Alcohol Clin Exp Res 1995; 19:510–516Crossref, Medline, Google Scholar

