
Asymmetrical Contribution of Brain Structures to Treatment-Resistant Depression As Illustrated by Effects of Right Subgenual Cingulum Stimulation
Mr. A’s first depressive episode was successfully treated with a tricyclic antidepressant, which was discontinued after 1 year. Mr. A remained symptom-free until he was 49 years old, when a second severe, nonpsychotic major depressive episode developed approximately 4 months after the detection and successful surgical resection of a localized prostate cancer. Symptoms were sustained (although with some fluctuations in severity lasting a few months) with little to no response to various treatments including paroxetine, fluoxetine, sertraline, mirtazapine, and clomipramine prescribed at maximal doses and taken for sufficient duration. Some of these antidepressants were augmented with lithium, divalproex sodium, risperidone, or olanzapine without benefit. Supportive psychotherapy was provided throughout the episode combined with a formal trial of cognitive behavior therapy (CBT), again without clinical benefit.
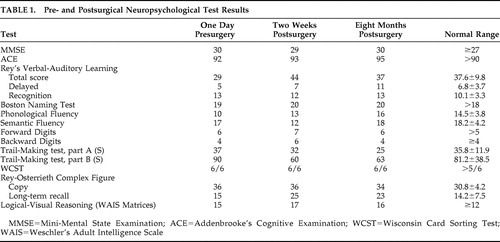
The patient was referred for consultation to our center after 10 years, at age 60. At the time of first evaluation, the patient had been unemployed for 3 years, was receiving disability benefits, and had experienced a loss of 17 kg in the previous 6 months. Neurological and medical examinations were normal. On mental status examination the patient appeared as a thin 60-year-old male, looking older than his stated age, disheveled, with poor self-care and grooming, no abnormal movements, and coherent but slow and impoverished speech. His thought content was focused on overvalued ideas of financial ruin. The patient was not suicidal. His mood was self-described as “horrible” and his affective expression was flat and congruent with the reported mood. He had no hallucinations in any sensory modality, Schneiderian first-rank symptoms, passivity experiences, obsessions, compulsions, phobias, depersonalization, or derealization. He retained insight into the nature of his health problem and his judgment was appropriate. A new medication regimen was instituted and included extended-release venlafaxine, 225 mg p.o. daily, and quetiapine, 200 mg p.o. twice daily, which remains unchanged to this day. A course of CBT was also initiated, but the patient was unable to comply with therapy demands due to poor attention and low energy.
Given continued severe symptoms, failure of standard and combination pharmacotherapy and psychotherapy, and absence of any neurological contraindications, ECT, a relatively seldom used treatment in his cultural setting, was considered for this patient. The patient underwent a series of 15 bilateral ECT sessions with seizure duration between 28 and 55 seconds. This ECT course brought about a mild and short-lived subjective amelioration; mood, initiative, sleep, and appetite modestly improved. The patient and his family deemed this improvement unsatisfactory.
Mr. A regressed to his previous level of symptom severity approximately 6 weeks after the last ECT session, in spite of continued treatment with venlafaxine, 225 mg per day, and quetiapine, 200 mg twice a day, as stated above. He had subjective complaints of attention and memory difficulties and these worsened transiently after the ECT course. Both the patient and his family felt the mild improvement in symptoms did not warrant a further course of ECT or maintenance ECT sessions. As recommended by a team meeting in the psychiatry department, the patient and his family were then offered deep-brain stimulation (DBS) of white matter immediately adjacent to Brodmann’s cortical area 25 (Cg25-DBS, subgenual cingulum) bilaterally as an experimental treatment for refractory depression that had shown promise in a recently published case series. 1 This procedure was offered instead of other neurosurgical options such as ablations or vagus nerve stimulation because of the extensive experience in our center (i.e., >200 interventions) with the use of DBS as a treatment of idiopathic Parkinson’s disease. Written consent was obtained from the patient and his wife. The protocol and consent form were modified from the original Canadian Cg25-DBS pilot study 1 and explicitly stated that the procedure was experimental, requiring testing of different stimulation parameters to optimize potential clinical effects and to minimize adverse events. Prior to the procedure, the approval by the local bioethics committee, the signed consent form, a summary of the patient’s history, and the technical description of the device were all reviewed—and approved—by the Argentine National Administration on Medications and Medical Devices (ANMAT), the national government agency regulating the local use of experimental drugs and medical devices. The patient was implanted as previously described. 1
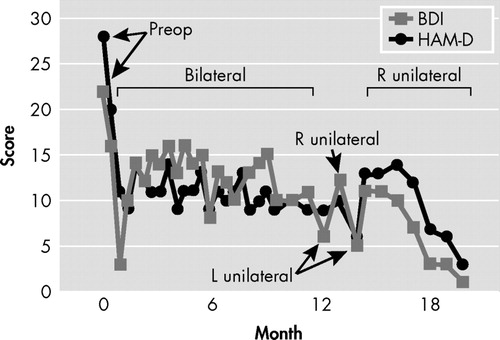
With the patient awake, different contact patterns were probed in the operating room, and some patterns consistently produced a sense of well-being that the patient described as “a relief of the tension in my neck.” After the operation, additional systematic testing of different pairs of bilateral and symmetrical contacts at varying current settings was performed in the first 3 days while in the hospital (contacts were changed every 3 hours during the daytime). Initial stimulation parameters were selected based on the Canadian pilot study 1 , 2 (voltage 1.5 V, pulse width 70 μsec, and frequency 90 Hz), and all these parameters were titrated upward to a maximum of 6 V, 90 μsec, and 130 Hz for each pair of electrodes at the aforementioned time. Testing of different combinations of contacts and currents continued in an outpatient setting, with evaluations and changes performed every 2 weeks. Response to each pattern of stimulation was based upon the subjective account offered by the patient while in the hospital, and then by means of the Beck Depression Inventory (BDI) and 21-item Hamilton Depression Scale (HAM-D), as depicted in Figure 1 . This process continued for 6 months with stimulation parameters in electrodes 2 (left hemisphere) and 6 (right hemisphere) associated with the most consistent symptom amelioration: frequency 120 Hz, pulsewidth 90 μsec, and voltage 4.5 V. After 6 months, the patient’s improvement reached a plateau, despite an initial and sustained drop in both Hamilton and BDI scores (see Table 1 , Figure 1 ). Further adjustments did not facilitate additional change in scores, which remained stable. As such, at 12 months and based on literature data suggesting a differential contribution of the left and right hemispheres to mood regulation, we began testing unilateral Cg25-DBS with those stimulation parameters found to be optimal in bilateral stimulation with the purpose of pursuing complete remission of depressive symptomatology. As shown in Figure 1 , left unilateral (L) stimulation appeared to be followed by rapid (i.e., less than 1 week) worsening of mood. At the next appointment (2 weeks later), Mr. A actually stated “by the time I reached the elevators, I was not feeling well already.” Even though, as depicted in Figure 1 , the patient did not return to his preoperative degree of depressive symptomatology, per his subjective account of mood, feelings of inner tension, and anergia, his status was distinctly different than experienced with bilateral stimulation, beginning within the hour of changing stimulation parameters. At this point, settings were switched to right unilateral stimulation (R, Figure 1 ), which, in contrast to left unilateral stimulation, resulted in not only reversal of the new symptoms but improvement beyond that seen with bilateral stimulation. With right unilateral stimulation, consistent remission of symptoms was seen within 2 weeks, both as rated by the patient with the BDI and as directly observed by the psychiatrist and quantified with the HAM-D. Mr. A was made aware by staff when stimulation parameters were being changed, but he remained blind to what these parameters were, and if stimulation was bilateral, right, or left. The psychiatrist who conducted the rating was not blind to these changes.
 |
Retesting of left unilateral stimulation performed with the purpose of ruling out a placebo effect or a temporary worsening unrelated to the type of stimulation resulted in similar transient worsening of symptoms within hours ( Figure 1 ) and similar accounts of deterioration in mood, anxiety, and lack of energy, whereas reinstitution of right unilateral stimulation brought about full symptom remission within 4 weeks that continues to the present time (now over 12 months later). No washout periods with stimulators off, or resumption of bilateral stimulation, were used when changing unilateral stimulation from one side to the other. No further trials were conducted because the patient remains fully remitted on unilateral right stimulation at 120 Hz, 90 μsec, and 4.5 V.
Overall, the surgical procedure and testing of all contacts at the various parameters was generally well tolerated. There were no behavioral or cognitive adverse events at any settings ( Table 1 ), and unilateral stimulation was not associated with cognitive symptoms. However, initial postoperative testing of the most proximal electrodes (bilateral stimulation, 120 Hz, 90 μsec, 3 V at contacts 3 and 7; i.e., those lying immediately adjacent to subcallosal cingulate gray matter) resulted in no change in mood symptoms but acute onset of orthostatic hypotension that normalized immediately when stimulation was discontinued. Such autonomic changes were seen at no other contacts. Retest of the deepest electrodes bilaterally 6 months postoperatively was performed because prior orthostatism could have been related to the postoperative status. With repeat testing, reversible asymptomatic, albeit significant (−15 mmHg), decrease in mean arterial pressure was induced within 60 seconds of stimulation onset. Unilateral stimulation using these contacts was not performed.
DISCUSSION
Major depressive disorder is the leading cause of disability-adjusted life years lost for people ages 15–44 years old worldwide. 2 Although the etiology of depression remains relatively obscure, the available evidence suggests that major depressive disorder is a complex disorder that results from an interaction of biological, genetic, psychosocial, and environmental factors. 3 Functional neuroimaging studies have broadened our understanding of how these complex factors impact the CNS to produce major depressive disorder symptoms 4 , 5 and how various treatments mediate recovery. 6 – 9 Bilateral deep brain stimulation of the white matter immediately beneath the subcallosal cingulate was in fact developed based upon the results of such studies, 1 and is currently being investigated as a potential treatment option for patients who fail to respond to approved methods for management of treatment-resistant depression. 10
A critical issue in the management of this case was the criteria used both to define treatment resistance and as a foundation for offering a potential new treatment of yet unproven efficacy. Treatment-resistant depression has been traditionally defined as an inadequate response to an adequate course of treatment in a patient with major depressive disorder. 11 Opinions vary regarding precisely what constitutes an inadequate response. Thase and Rush 12 proposed a five-stage system for defining treatment-resistant depression, which ranges from lack of response to a single adequate trial of antidepressants to lack of response to ECT, the most effective single treatment for depression. 13 , 14 Reported prevalence rates for treatment-resistant depression vary according to which criterion is used to define the condition and the clinical setting in which the study was carried out, with progressively higher rates occurring in ambulatory settings, psychiatric hospitals, and tertiary care settings. Our patient would clearly meet Stage 5 treatment-resistant depression, defined as failure to respond to three antidepressants and failure to respond to ECT. 11 This condition, as exemplified by our patient, is associated with a remarkable burden in direct and indirect costs, in loss of quality of life for patients and their families, and in increased risk for suicide and substance abuse. 15 – 17 While multiple factors are known to contribute to treatment nonresponse among individuals with major depressive disorder, the two most common causes—poor adherence to prescribed treatment 18 and poor tolerability 19 , 20 —were not at issue here. Chronicity of depression (in terms of duration of the present episode and number of previous episodes), older age, psychiatric and medical comorbidities, and symptom severity are all associated with delayed time to remission 12 , 21 , 22 and were serious considerations in this case. It is well recognized that medical and psychiatric comorbidities can play a crucial role in treatment resistance. In STAR*D, 53% of patients had one or more significant medical comorbidities. Older age, lower socioeconomic and educational status, and lack of a family history of depression were additionally associated with this complicating factor 23 and contributed to treatment resistance. In this case, there were no such comorbid factors except for the inactive prostate cancer.
The steps taken in the treatment of Mr. A when his depressive symptoms failed to respond to efficacious treatments reflect the usual practice in the management of treatment-resistant depression, though, as noted above, there is a remarkable paucity of well-controlled and adequately powered studies comparing different treatment options to manage advanced-stage treatment-resistant depression. 11 Increasing antidepressant dose switching or combining with a second class of antidepressant, augmentation strategies (with lithium, T3, or an atypical antipsychotic), and an adequate course of ECT are the usual steps to manage treatment-resistant depression and were tried in succession with Mr. A, albeit unsuccessfully. Mr. A’s case is an example of a significant number of patients who remain seriously ill despite currently available approaches to treatment-resistant depression. A major shortcoming in the treatment of depression in general, but in treatment-resistant depression specifically, is the lack of predictors of response to particular treatments in individual patients. 24 In the last decade, functional brain imaging techniques have begun to delineate the putative brain circuits that are altered in depression, and the functional brain correlates of antidepressant treatment nonresponse. 25 – 27 These research efforts have prompted the design of interventions that attempt direct modulation of abnormal circuits for alleviation of otherwise refractory depressive symptoms. 5
Brain Circuits Involved in Depression and Deep Brain Stimulation of Brodmann’s Area 25
A brain circuit model explaining the development, response to treatment, and neurobiological basis of treatment nonresponse in depression maintains the classical neurological tradition of symptom localization, initially using lesion-deficit correlational analyses. 5 Before the advent of widely available functional brain imaging techniques, the study of the effect of discrete brain lesions helped to delineate areas putatively involved in the onset of depressive symptoms. With the help of those studies, the prefrontal cortex (PFC) and different cerebral structures interconnected with it have long been recognized as key brain areas in the pathogenesis of depression. The last decade witnessed a myriad of studies that used functional brain imaging techniques with increasingly better anatomical resolution to define the role of cortical and subcortical brain areas involved in the development, maintenance, response to treatment, and treatment-refractoriness of depressive illness. 5 – 9 , 26 , 27
Concordant with the results of lesion studies, resting-state blood flow and glucose metabolism measures using PET commonly reported frontal cortex and cingulate abnormalities, especially decreased function. 8 , 28 Within the frontal lobe, the most consistent abnormalities have been found in ventrolateral and dorsolateral prefrontal cortex (Brodmann’s areas 9, 10, 46, and 47), as well as orbitofrontal and ventromedial PFC (Brodmann’s areas 10, 11, and 32). Less consistent abnormalities have been reported for limbic and subcortical structures, including amygdala, anterior temporal and insular cortices, and sections of the basal ganglia and thalamus. 5 Both bilateral and asymmetrical findings have been reported. Although some functional neuroimaging findings are consistent, others display significant interindividual variability, possibly related to differences in genetic predisposition, early life adverse experiences, heterogeneity in clinical presentation, illness severity, and cognitive dysfunction. A replicated observation in these resting-state studies has been the inverse relationship between PFC activity and depression severity. Brody and coworkers 29 have proposed a more complex relationship between behavioral abnormalities and cerebral activity in depression, in the form of a ventral-dorsal segregation of PFC functions. In this view, anxiety/tension is positively correlated with ventral PFC activity, and psychomotor and cognitive slowing are negatively correlated with dorsolateral PFC activity, findings supported in viewing brain imaging studies that compare major depression and generalized anxiety disorder. 30
Functional neuroimaging studies that explore the neurobiological basis of response and refractoriness to diverse treatments for depression are of particular importance in the definition of potential targets for direct modulation of cerebral structures in patients with treatment-resistant depression. Resting-state PET studies and fMRI using behavioral tests have consistently reported that increased pretreatment activity in the pregenual cingulate cortex (aCg24; Brodmann’s area 24) distinguishes responders to a series of antidepressant interventions from nonresponders. 5 , 31 – 33 In contrast, subgenual cingulate cortex (Cg25, Brodmann’s area 25) hyperactivity has been shown to predict response to sleep deprivation 7 and cingulotomy 34 in patients who had previously not responded to antidepressants, thus constituting a potential neurobiological marker of treatment-resistant depression. 35
Mayberg 5 recently elaborated on a previously proposed multinode circuit model of clinical depression that evolved mainly from the functional neuroimaging findings summarized above. In this model, regions of potential involvement are grouped in four major nodes with different roles in the onset and maintenance of, but also in response to treatment of, depressive symptoms ( Figure 2 ). In this model, two of the clusters are mostly concerned with interoceptive phenomena and the somatic correlates of depression ( Figure 2 , light gray panels), and the other two clusters are concerned with exteroception and neocortical processing and their abnormalities during depressive episodes ( Figure 2 , dark gray panels). As consistently observed across studies, commencing with early functional neuroimaging reports, reciprocal interactions between ventral limbic—“interoceptive”—and dorsal neocortical—“exteroceptive”—regions are characterized by relative hyperactivity of the former and relative hypoactivity in the latter. 5 Medial PFC, including orbitofrontal structures, are considered separately in the model to account for their distinctive role in the cognitive control of emotional status. Similarly, subcortical structures participating in cortical-limbic loops involved in emotion, including amygdaloid nuclei, ventral tegemental area, thalamic nuclei, and ventral striatal regions, have been segregated from anterior insula and especially the hypothalamus, given their role in the processing of novel stimuli and salience as well as in reinforcement and therefore learning. 5 , 36 – 40 The proposed model provides a conceptual framework to understand the rationale for using a series of DBS targets in depression. Thus, in addition to Cg25, other targets modulating cortical-limbic circuits presumably abnormal in depression have been proposed, including anterior limb of the internal capsule, nucleus accumbens, inferior thalamic peduncle, rostral cingulate gyrus, and left forebrain activation via vagus nerve stimulation. 10 , 39 – 43 However, the model does not incorporate emerging data on an asymmetrical role of brain hemispheres in mood regulation, as illustrated by the case of Mr. A.

ACg=anterior cingulate cortex; a-ins=anterior insula; amg=amygdala; BS=septal nuclei; Cg=cingulate cortex; dm-Th=dorsomedial thalamus; dp-HC=dorsal-posterior hippocampus; hth=hypothalamus; MCg=medial cingulate cortex; mF=medial frontal cortex; mOF=medial orbital frontal cortex; Par=parietal cortex; PFC=prefrontal cortex; PM=premotor cortex; va-HC=ventral anterior hippocampus; vta=ventral tegmental area; vst-cd=ventral striatum-caudate. Numbers designate cortical Brodmann’s areas.
Asymmetrical Contribution of Brain Structures to the Development of Depressive Symptoms and Response to Treatment
The asymmetrical response of Mr. A to subcallosal cingulate DBS suggests a differential contribution of left and right prosencephalic structures in mood regulation. Structural and functional asymmetry of the human brain has been recognized since the second half of the 19th century due to the pioneering work of Dax, 44 Broca, 45 and Wernicke 46 on the preeminent role of left-sided lesions in the production of most cases of aphasia. An additional landmark in the study of brain asymmetry was the work of Tseng and Sperry 47 and their collaborators with patients who had undergone surgical transection of their corpus callosum to prevent the propagation of seizures. This work extended the concept of brain asymmetry to “higher” cognitive functions beyond language. Studies on hemi-neglect in patients with right-sided lesions of the brain revealed a significant asymmetry in the neural circuits involved in the regulation of attention to outside events, which involves not only a series of sensory modalities but motivational and emotional responses to them as well. 48 However, knowledge of the brain’s functional asymmetries and their impact on emotion and behavior is limited, in part, due to the fact that good experimental models have been lacking until recently. This is likely, in part, due to the view that lateralization of brain functions and behavior has long been considered a purely human trait. 49 The seminal work of Nottebohm and colleagues 50 , 51 on asymmetries in the neural control of song production in birds has modified this perception. Subsequently, asymmetric contributions of brain hemispheres to a variety of behaviors have been recognized as a ubiquitous feature of CNS organization across species, including fish, birds, amphibians, rodents, and nonhuman primates, 52 though themes relative to “higher” cognitive functioning have been more intensively studied than asymmetric contributions of brain hemispheres to emotion, autonomic/visceral sensation, and mood and motivated behavior.
Nonetheless, hemispheric laterality has been a preeminent theme in the field of early lesion correlation with depression (i.e., whether left and right cerebral structures contribute differently to dysregulation of mood and the emergence of depressive symptoms). A series of studies supports the hypothesis of an asymmetrical role of the two hemispheres. The majority of data showing a differential role of left versus right CNS circuits in the generation of depressive symptoms were derived from studies investigating incident depression after a localized cerebral lesion, especially a stroke. 53 – 56 These studies offered evidence that left (but not right) anterior hemisphere lesions were causally associated with depression, which was interpreted as suggesting that dysfunction of left prefrontal circuits might be a feature in this disorder.
An alternative explanation, in light of more recent studies on brain function as described below, could be that right PFC activity might become predominant after suffering left-sided lesions, and this imbalance instead of left PFC dysfunction might underlie the development of depressive symptoms. Early positron emission tomography (PET) and electroencephalographic (EEG) studies suggested a pronounced left PFC hypoactivity 57 , 58 as well. However, a series of attempts at replication, as well as meta-analyses, do not support a systematic effect of lesion location on poststroke depression. 59 – 61 Koenigs and Grafman 62 recently reported on the association between left, right, or bilateral cerebral lesions and depressive symptoms 15 and 35 years after the index brain injury in a cohort of male Vietnam War veterans. The demographic homogeneity of the sample permitted excellent comparability between groups and in addition with a group of veterans who had not sustained significant brain damage. The study concluded that in this population in particular there were no associations between depressive symptoms and side of the cerebral lesion. 62 Taken together, studies on the correlation between discrete brain lesions and depression have not clearly demonstrated a uniform role of hemispheric laterality in the appearance of depressive symptoms.
Findings of altered bilateral activity of components of circuits involved in depression have been observed, but other studies strongly suggest an asymmetry in the contribution of anterior cortical-limbic circuits to the appearance of depressive symptoms. Dougherty et al. 34 attempted to define potential resting-state cerebral activity predictors of response to cingulotomy, using [ 18 F]fluorodeoxyglucose (FDG)-PET. The study is of particular interest because the patients belonged, like Mr. A, to a population with the most severe and most treatment-refractory forms of major depressive disorder. They observed that hyperactivity in the left subgenual cerebral cortex and left thalamus correlated with improvement in depressive symptom severity following cingulotomy. 34 In a study exploring the relationship between changes in cerebral metabolic activity and improvement of depressive symptoms after sleep deprivation, Wu et al. 7 observed that improvement in depressive symptomatology was correlated with an increase in the activity of the left temporal cortex and a decrease in left anterior medial PFC, without similar contralateral findings. In a recent study, Konarsky et al. 27 detected an increase in glucose metabolism in Brodmann’s areas 24 and 32, the interphase sector between “cognitive” and “visceral/emotional” portions of the cingulate cortex, specifically on the left side, in depressed patients who had not responded to cognitive behavior therapy (CBT) or venlafaxine treatment.
Other evidence points to a preeminent role of right cortico-subcortical functioning in the modulation of mood and in response to antidepressant treatment. Kennedy et al. 63 described changes in brain glucose metabolism associated with 16-week-long treatment either with venlafaxine or CBT. Right subgenual/ventromedial frontal increase in metabolism was observed in patients responding to CBT, whereas decreases in metabolism in the right posterior subgenual cingulate were associated with response to venlafaxine. Notably, reduction in metabolic activity bilaterally in the orbitofrontal cortex was associated with response to either treatment modality. In contrast, decreased metabolism restricted to the left orbitofrontal cortex was observed irrespective of treatment group or treatment outcome (i.e., left hemispheric changes were irrelevant to the response of depressive symptoms). The Kennedy et al. 63 study extends asymmetrical brain metabolic findings to the right thalamus, a subcortical structure that is a component of the circuit involved in mood regulation. These findings, as well as the fact that Mr. A responded to unilateral right stimulation of Brodmann’s area 25, are also congruent with the observation that the right dorsomedial PFC is activated relatively selectively in a wide range of emotional tasks, including recollection of affectively meaningful life events, 64 attention to subjective feelings, 65 and processing of emotion-related meanings. 66 In this fMRI study, healthy subjects were presented a series of pictures combined with relevant captions resulting in positive, negative, and neutral affective connotations. Presentation of negative picture-caption pairs was associated with activation of a series of cortical and subcortical structures, in all cases localized to the right hemisphere: medial and right middle frontal gyri (Brodmann’s area 9), right anterior cingulate gyrus (areas 24 and 32), and right thalamus. 66
Our provisional observation of prolonged depression remission with right, but not left, unilateral stimulation of the subgenual cingulate cortex (Brodmann’s area 25) is concordant with those findings in response to antidepressants and the induction of negative affect in healthy individuals, and suggests that efficacy of DBS in Mr. A was entirely explained by its effects on the right cerebral hemisphere. Some of the implicated right-sided structures are in the vicinity of the midline, so the efficacy of right DBS might also be explained, at least in part, by bilateral effects of a single electrode placed on that side. This is best appreciated by observing the trajectory of tracts in the vicinity of—and thus presumably affected by—the DBS electrodes in Mr. A ( Figure 3 ). As seen in this figure, neuronal tracts affected by each electrode show ipsilateral projections to the PFC. Tracts neighboring the right electrode, however, seem to additionally project contralaterally to a greater extent than those in the vicinity of the left electrode in this patient. This emphasizes the need to further define the tracts critical for mood regulation 67 and perhaps suggests that, in the future, attempts at regulating brain circuits might need to be “customized” to match the interindividual variability in anatomical projections. This approach has already been proposed for subthalamic nuclei DBS in idiopathic Parkinson’s disease. 68 Regardless of the reasons for the present observation of asymmetric contribution of right versus left DBS to the amelioration of depressive symptoms, Mr. A’s case probably points to a need to take into account hemispheric asymmetries in models of depressive symptom generation.
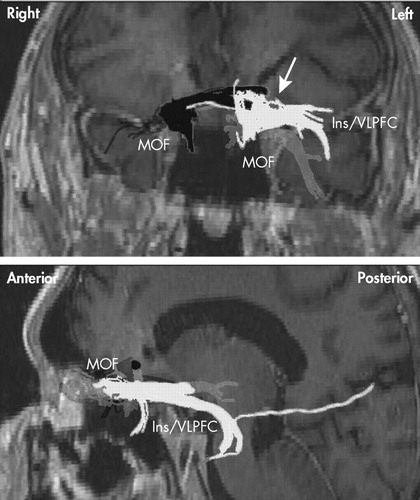
Shown in dark gray are white matter tracts passing through right-sided deep brain stimulation targets. Note that there are ipsilateral connections to the medial orbital frontal cortex from each side. Only the right-sided contact shows contralateral tract involvement (white arrow). Notable also are the more lateral tracts to the lateral prefrontal and anterior insula from the left-sided contact (in light gray). MOF=Medial orbital frontal cortex; Ins=Insula; VLPFC=Ventral lateral prefrontal cortex.
If further studies confirm, that just as is the case with attention, 48 the right hemisphere serves a preeminent role in normal mood regulation and clinical depression, the question arises whether lateralization applies to all components of Mayberg’s model of depression delineated above 5 , 25 or if instead “interoceptive” (subcortical/paleocortical) or “exteroceptive” (neocortical) nodes are preferentially lateralized in their contribution to mood regulation.
Asymmetrical Central Representation of Visceral Function and Its Relationship With Regulation of Mood and Emotion
There is recent evidence favoring lateralization in the control of abnormal visceral sensation in the context of mood dysregulation. Coen et al. 69 described the patterns of brain activation during the processing of painful and nonpainful visceral stimuli (esophageal distention) in different affective states. Evidence for a dominant role of the right hemisphere in the bodily correlates of negative emotion was obtained that specifically involves the right anterior cingulate (Brodmann’s areas 24 and 32), right anterior insula, and inferior frontal gyrus. 69 Gut sensations evoking increased heart rate, low-frequency/high-frequency heart rate variability ratio, and plasma epinephrine, an autonomic pattern characteristically associated with depression, 70 – 73 are associated with increased activation of right insula, right orbitofrontal cortex, and right parahippocampal gyrus, as well as subcortical and brainstem structures. 74 This further suggests a right hemispheric dominance in the control of visceral sensation associated with negative mood and the autonomic profiles characteristic of it.
Induction of negative mood has been associated with hyperactivity of right insula and prefrontal cortical structures in healthy volunteers as well. 4 Consistent with this view, Giesecke et al. 75 observed that symptoms of depression correlate with pain-evoked activation of the right anterior insula. Craig 76 , 77 summarized evidence explaining the lateralization of emotion processing and suggested that the right insular cortex might be the principal CNS structure responsible for sentience and self-consciousness. He proposed that prosencephalic emotional asymmetry in turn reflects an asymmetrical representation of homeostatic activity, ultimately explained by asymmetries in the autonomic nervous system hierarchy including peripheral sympathetic and parasympathetic structures. Vagal afferents innervate the nucleus of the solitary tract, whereas sympathetic afferents terminate in lamina I of the dorsal horn of the spinal cord. From there, axons innervate the basal (parasympathetic) and posterior (sympathetic) portions of the ventromedial nucleus of the thalamus. This topographical organization is present in primates, and is extremely well-developed only in humans. 76 , 78 – 80 Ultimately, the left anterior insula is activated by afferent information related to parasympathetic functions, whereas the right anterior insula is activated by afferent information associated with sympathetic activity (e.g., pain). 76 Thus, activity in the right prosencephalon is associated with energy expenditure, sympathetic activity, arousal, aversive behavior, and survival emotions, whereas activity in the left forebrain is associated with energy nourishment, parasympathetic activity, relaxation, appetitive behavior, and affiliative emotions. 77 There is, in fact, a growing body of evidence that suggests that left and right forebrain structures are associated with positive and negative affect, respectively. 81 It has been suggested that this, in turn, reflects asymmetries in autonomic afferent input onto limbic structures. 82 , 83 The concept behind the correlation between anatomical segregation of afferent autonomic input and cerebral involvement in the processing of emotions is the cornerstone of the James-Lange theory, which maintains that emotional feeling states are initiated by autonomic afferent information reaching consciousness. 84 , 85 This theory has received support recently because of evidence that peripheral autonomic clues are crucial for motivated behavior. 86 , 87
Complete suppression of otherwise treatment-refractory depressive symptoms in Mr. A by means of right DBS (presumably blocking neural transmission in forebrain limbic structures on that side 88 ) is consistent with this growing body of evidence on lateralization of emotional regulation and also with preliminary observations that another neuromodulation tool useful in treatment-resistant depression, vagus nerve stimulation, might exert its beneficial effect via activation of left forebrain structures and reciprocal inhibition of right forebrain structures. 39 , 76 Although we do not have functional brain imaging studies available to demonstrate the effects of DBS on either side of the brain in Mr. A, we have collected data on cardiac autonomic activity in resting conditions ( Figure 4 ). Using analysis of heart rate variability, 89 we identified high-frequency (parasympathetic) and low-frequency (baroreflex, but predominantly sympathetic) influences on the sinus node (Biomedical Signal Analysis Group, Department of Applied Physics, University of Kuopio, Finland). As shown in Figure 4 , unilateral right DBS was characterized by remarkably lower low-frequency/high-frequency ratio (an estimation of the sympathovagal balance on the heart) 89 , 90 than unilateral left DBS. As expected, heart rate was higher in the latter condition, further suggesting increased sympathetic and/or decreased vagal input to the heart. If confirmed, these observations suggest that emotional well-being depends not only on “top-down” but also “right-left” modulation of prosencephalic activity, and would permit reconceptualization of older studies on the effect of brain lesions in the production of depressive symptoms. The common observation of left lesions being associated with the development of depression might, in light of this model, be explained less by the interruption of brain circuits on the left side than by unopposed influences of right forebrain and limbic structures, physiologically associated with negative affect, sympathetic activity, and energy expenditure. Stimulation of the unencumbered right hemisphere, resulting in inhibition of local and remote structures within these circuits, 1 may help to restore interhemispheric balance.
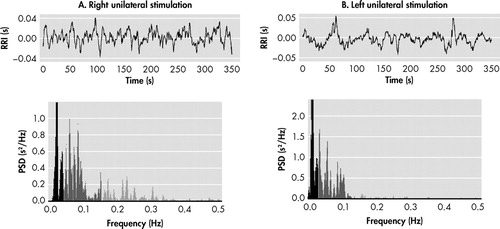
The top panels depict the duration of successive cardiac beats (RR intervals, s), and the bottom panels show the fast Fourier transform analysis of the resulting signals (power spectral density, PSD, s 2 /Hz) of resting-state recordings obtained during unilateral right (A) and unilateral left (B) stimulation of the subgenual cingulum. Low-frequency (LF) heart rate variability (0.03–0.15 Hz, light gray) reflects baroreflex but predominantly sympathetic activity. High-frequency (HF) heart rate variability (0.15–0.4 Hz, light gray) measures respiratory sinus arrhythmia and is therefore a specific indicator of cardiac parasympathetic activity. LF/HF ratio, a measure of sympathovagal balance onto the heart, 88,89 was higher during left than during right stimulation (8.17 vs 2.42, respectively). Most noticeable is the withdrawal of vagal activity during left stimulation (0.15–0.4 Hz range). Accordingly, heart rate was higher in this condition (100.4 bpm vs 90.2 bpm during right stimulation).
Summary and Recommendations for Future Research
Although still relatively small in number, there is burgeoning evidence of the efficacy of DBS in the treatment of the most severely treatment-refractory depressed patients. 10 , 41 The current case adds to this database, and like previous reports it demonstrates the potential utility of this intervention in those patients who have failed to respond to past adequate treatment with psychotherapy, a variety of antidepressants, and ECT. Although the present observation needs to be confirmed in double-blind studies, as Mr. A’s psychiatrist was not blind to changes in stimulation parameters, this case raises important questions about laterality of mood regulation and unilateral versus bilateral DBS treatment, which warrant further study. The present case example and the bulk of evidence highlight the preeminence of cortical and subcortical structures in the right hemisphere in the generation and maintenance of depression. This hypothesis remains to be confirmed with studies specifically addressing this question, including animal models of the disease as appropriate.
1. Mayberg HS, Lozano AM, Voon V, et al: Deep brain stimulation for treatment-resistant depression. Neuron 2005; 45:651–660Google Scholar
2. World Health Organization: WHO Global Burden of Disease, 2004 Update, Part 3: Disease incidence, prevalence and disability. Available at www.who.int/healthinfo/global_burden_disease/GBD_report_2004update_part3.pdfGoogle Scholar
3. Gillespie CF, Garlow SJ, Schatzberg AF, et al: Neurobiology of mood disorders, in Psychopharmacology, 4th ed. Edited by Shatzberg AF, Nemeroff CB. Washington, DC, American Psychiatric Publishing, 2009, pp 903–944Google Scholar
4. Mayberg HS, Liotti M, Brannan SK, et al: Reciprocal limbic- cortical function and negative mood: converging PET findings in depression and normal sadness. Am J Psychiatry 1999; 156:675–682Google Scholar
5. Mayberg HS: Targeted electrode-based modulation of neural circuits for depression. J Clin Investigation 2009; 119:717–725Google Scholar
6. Little JT, Ketter TA, Kimbrell TA, et al: Venlafaxine or bupropion responders but not nonresponders show baseline prefrontal and paralimbic hypometabolism compared with controls. Psycopharmacol Bull 1996; 32:629–635Google Scholar
7. Wu J, Buchsbaum MS, Gillin JC, et al: Prediction of antidepressant effects of sleep deprivation by metabolic rates in the ventral anterior cingulated and medial prefrontal cortex. Am J Psychiatry 1999; 156:1149–1158Google Scholar
8. Mayberg HS: Modulating dysfunctional limbic-cortical circuits in depression: towards development of brain-based algorithms for diagnosis and optimised treatment. Br Med Bull 2003; 65:193–207Google Scholar
9. Pizzagalli D, Pascual-Marqui RD, Nitschke JB, et al: Anterior cingulate activity as a predictor of degree of treatment response in major depression: evidence from brain electrical tomography analysis. Am J Psychiatry 2001; 158:405–415Google Scholar
10. Lozano AM, Mayberg HS, Giacobbe P, et al: Subcallosal cingulate gyrus deep brain stimulation for treatment-resistant depression. Biol Psychiatry 2008; 64:461–467Google Scholar
11. Nemeroff CB: Prevalence and management of treatment-resistant depression. J Clin Psychiatry 2007; 68:17–25Google Scholar
12. Thase ME, Rush AJ: When at first you don’t succeed: sequential strategies for antidepressant nonresponders. J Clin Psychiatry 1997; 58:23–29Google Scholar
13. Janicak PG, Davis JM, Gibbons RD, et al: Efficacy of ECT: a meta-analysis. Am J Psychiatry 1985; 142:297–307Google Scholar
14. The UK ECT Review Group: Efficacy and safety of electroconvulsive therapy in depressive disorders: a systematic review and meta-analysis. Lancet 2003; 361:799–808Google Scholar
15. Greenberg PE, Stiglin LE, Finkelstein SN, et al: The economic burden of depression in 1990. J Clin Psychiatry 1993; 54:405–418Google Scholar
16. Crown WH, Finkelstein S, Berndt ER, et al: The impact of treatment-resistant depression on health care utilization and costs. J Clin Psychiatry 2002; 63:963–971Google Scholar
17. Russell JM, Hawkins K, Ozminkowski RJ, et al: The cost consequences of treatment-resistant depression. J Clin Psychiatry 2004; 65:341–347Google Scholar
18. Katon WJ, Von Korff M, Lin EH, et al: The pathways study: a randomized trial of collaborative care in patients with diabetes and depression. Arch Gen Psychiatry 2004; 61:1042–1049Google Scholar
19. Trivedi MH, Fava M, Wisniewski SR, et al, STAR*D Study Team: Medication augmentation after the failure of SSRIs for depression. N England J Med 2006; 354:1243–1252Google Scholar
20. Souery D, Amsterdam J, de Montigny C, et al: Treatment resistant depression: methodological overview and operational criteria. Eur Neuropsychopharmacol 1999; 9:83–91Google Scholar
21. Nierenberg AA, Wright EC: Evolution of remission as a new standard in the treatment of depression. J Clin Psychiatry 1999; 60:7–11Google Scholar
22. Thase ME: Introduction: defining remission in patients treated with antidepressants. J Clin Psychiatry 1999; 60:3–6Google Scholar
23. Yates WR, Mitchell J, Rush AJ, et al: Clinical features of depressed outpatients with and without co-occurring general medical conditions in STAR*D. Gen Hosp Psychiatry 2004; 26:421–429Google Scholar
24. Insel TR: Disruptive insight, in psychiatry: transforming a clinical discipline. J Clin Invest 2009; 119:700–705Google Scholar
25. Mayberg HS, Brannan SK, Mahurin RK, et al: Cingulate function in depression: a potential predictor of treatment response. Neuroreport 1997; 8:1057–1061Google Scholar
26. Siegle GJ, Carter CS, Thase ME: Use of fMRI to predict recovery from unipolar depression with cognitive behavior therapy. Am J Psychiatry 2006; 163:735–738Google Scholar
27. Konarski JZ, Kennedy SH, Segal ZV, et al: Predictors of nonresponse to cognitive behavioral therapy or venlafaxine using glucose metabolism in major depressive disorder. J Psychiatry Neurosci 2009; 34:175–180Google Scholar
28. Drevets WC, Savitz J, Trimble M: The subgenual anterior cingulate cortex in mood disorders. CNS Spectr 2008; 13:663–681Google Scholar
29. Brody AL, Saxena S, Mandelkern MA, et al: Brain metabolic changes associated with symptom factor improvement in major depressive disorder. Biol Psychiatry 2001; 50:171–178Google Scholar
30. Martin EI, Ressler KJ, Binder E, et al: The neurobiology of anxiety disorders: brain imaging, genetics, and psychoneuroendocrinology. Psychiatr Clin North Am 2009; 32:549–575Google Scholar
31. Pizzagalli DA, Oakes T, Fox A, et al: Functional but not structural subgenual prefrontal cortex abnormalities in melancholia. Mol Psychiatry 2004; 9:325, 395–405Google Scholar
32. Chen CH, Ridler K, Suckling J, et al: Brain imaging correlates of depressive symptom severity and predictors of symptom improvement after antidepressant treatment. Biol Psychiatry 2007; 62:407–414Google Scholar
33. Salvadore G, Cornwell BR, Colon-Rosario V, et al: Increased anterior cingulate cortical activity in response to fearful faces: a neurophysiological biomarker that predicts rapid antidepressant response to ketamine. Biol Psychiatry 2009; 65:289–295Google Scholar
34. Dougherty DD, Weiss AP, Cosgrove GR, et al: Cerebral metabolic correlates as potential predictors of response to anterior cingulotomy for treatment of major depression. J Neurosurg 2003; 99:1010–1017Google Scholar
35. Greicius MD, Flores BH, Menon V, et al: Resting-state functional connectivity in major depression: abnormally increased contributions from subgenual cingulate cortex and thalamus. Biol Psychiatry 2007; 62:429–437Google Scholar
36. Myers KM, Davis M: Mechanisms of fear extinction. Mol Psychiatry 2007; 12:120–150Google Scholar
37. Knutson B, Greer SM: Anticipatory affect: neural correlates and consequences for choice. Philos Trans R Soc Lond Biol Sci 2008; 363:3771–3786Google Scholar
38. Santini E, Quirk GJ, Porter JT: Fear conditioning and extinction differentially modify the intrinsic excitability of infralimbics neurons. J Neurosci 2008; 28:4028–4036Google Scholar
39. Devous MD, Husain M, Harris TS, et al: Effects of VNS on regional cerebral blood flow in depressed subjects. Eur Psychiatry 2002; 17:113–114Google Scholar
40. Schlaepfer TE, Cohen MX, Frick C, et al: Deep brain stimulation to reward circuitry alleviates anhedonia in refractory major depression. Neuropsychopharmacology 2008; 33:368–377Google Scholar
41. Malone DA Jr, Dougherty DD, Rezai AR, et al: Deep brain stimulation of the ventral capsule/ventral striatum for treatment-resistant depression. Biol Psychiatry 2009; 65:267–275Google Scholar
42. Jiménez F, Velasco F, Salin-Pascual R, et al: A patient with a resistant major depression disorder treated with deep brain stimulation in the inferior thalamic peduncle. Neurosurgery 2005; 57:585–593Google Scholar
43. Sakas DE, Panourias IG: Rostral cingulate gyrus: a putative target for deep brain stimulation in treatment-refractory depression. Med Hypotheses 2006; 66:491–494Google Scholar
44. Cubelli R, Montagna CG: A reappraisal of the controversy of Dax and Broca. J Hist Neurosci 1994; 3:215–226Google Scholar
45. Broca PP: Perte de la parole, ramollissement chronique et destruction partielle du lobe antérieur gauche du cerveau. Bulletin de la Société Anthropologique 1861; 2:235–238Google Scholar
46. Wernicke C: Der aphasische Symptomencomplex: Eine psychologische Studie auf anatomischer Basis. Breslau M Crohn und Weigert, 1874Google Scholar
47. Teng EL, Sperry RW: Interhemispheric rivalry during simultaneous bilateral task presentation in commissurotomized patients. Cortex 1974; 10:111–120Google Scholar
48. Mesulam MM: Spatial attention and neglect: parietal, frontal, and cingulate contributions to the mental representation and attentional targeting of salient extrapersonal events. Philos Trans R Soc Lond B Biol Sci 1999; 354:1325–1346Google Scholar
49. Tommasi L: Mechanisms and functions of brain and behavioral asymmetries. Philos Trans R Soc Lond B Biol Sci 2009; 364:855–859Google Scholar
50. Nottebohm F: Neural lateralization of vocal control in a passerine bird. I. Song. J Exp Zool 1971; 177:229–261Google Scholar
51. Nottebohm F, Arnold AP: Sexual dimorphism in vocal control areas of the songbird brain. Science 1976; 194:211–213Google Scholar
52. Bisazza A, Dadda M, Facchin L, et al: Artificial selection on laterality in the teleost fish girardinus falcatus. Behav Brain Res 2007; 178:29–38Google Scholar
53. Robinson RG, Kubos KL, Starr LB, et al: Mood disorders in stroke patients: importance of location of lesion. Brain 1984; 107:81–93Google Scholar
54. Parikh RM, Lipsey JR, Robinson RG, et al: A two years longitudinal study of poststroke mood disorders: prognostic factors related to one and two year outcome. Int J Psychiatry Med 1988; 18:45–56Google Scholar
55. Shimoda K, Robinson RG: The relationship between poststroke depression and lesion location in long-term follow-up. Biol Psychiatry 1999; 45:187–192Google Scholar
56. Narushima K, Kosier JT, Robinson RG: A reappraisal of poststroke depression, intra- and inter-hemispheric lesion location using meta-analysis. J Neuropsychiatry Clin Neurosci 2003; 15:422–430Google Scholar
57. Henriques JB, Davidson RJ: Left frontal hypoactivation in depression. J Abnorm Psychol 1991; 100:535–545Google Scholar
58. Martinot JL, Hardy P, Feline A, et al: Left prefrontal glucose hypometabolism in the depressed state: a confirmation. Am J Psychiatry 1990; 147:1313–1317Google Scholar
59. Singh AA, Herrmann N, Black SE: The importance of lesion location in poststroke depression: a critical review. Can J Psychiatry 1998; 43:921–927Google Scholar
60. Carson AJ, MacHale S, Allen K, et al: Depression after stroke and lesion location: a systematic review. Lancet 2000; 356:122–126Google Scholar
61. Yu L, Liu CK, Chen JW, et al: Relationship between poststroke depression and lesion location: a meta-analysis. Kaohsiung J Med Sci 2004; 20:372–380Google Scholar
62. Koenigs M, Grafman J: Prefrontal asymmetry in depression? The long-term effect of unilateral brain lesions. Neurosci Lett 2009; 459:88–90Google Scholar
63. Kennedy SH, Konarski JZ, Segal ZV, et al: Differences in brain glucose metabolism between responders to CBT and venlafaxine in a 16-week randomized controlled trial. Am J Psychiatry 2007; 164:778–788Google Scholar
64. Reiman EM, Lane RD, Ahern GL, et al: Neuroanatomical correlates of externally and internally generated human emotion. Am J Psychiatry 1997; 154:918–925Google Scholar
65. Lane RD, Reiman EM, Ahern GL, et al: Neuroanatomical correlates of happiness, sadness, and disgust. Am J Psychiatry 1997; 154:926–933Google Scholar
66. Teasdale JD, Howard RJ, Cox SG, et al: Functional MRI study of the cognitive generation of affect. Am J Psychiatry 1999; 156:209–215Google Scholar
67. Johansen-Berg H, Gutman DA, Behrens TE, et al: Anatomical connectivity of the subgenual cingulate region targeted with deep brain stimulation for treatment-resistant depression. Cereb Cortex 2008; 18:1374–1383Google Scholar
68. McIntyre CC, Frankenmolle AM, Wu J, et al: Customizing deep brain stimulation to the patient using computational models. Conf Proc IEEE Eng Med Biol Soc 2009; 2009:4228–4229Google Scholar
69. Coen SJ, Yágüez L, Aziz Q, et al: Negative mood affects brain processing of visceral sensation. Gastroenterology 2009; 137:253–261Google Scholar
70. Guinjoan SM, Bernabo L, Cardinali DP: Cardiovascular tests of autonomic function and sympathetic skin responses in patients with major depression. J Neurol Neurosurg Psychiatry 1995; 58:299–302Google Scholar
71. Guinjoan SM, de Guevara MS, Correa C, et al: Cardiac parasympathetic dysfunction related to depression in older adults with acute coronary syndromes. J Psychosom Res 2004; 56:83–88Google Scholar
72. Guinjoan SM, Castro MN, Vigo DE, et al: Depressive symptoms are related to decreased low-frequency heart rate variability in older adults with decompensated heart failure. Neuropsychobiology 2007; 55:219–224Google Scholar
73. Carney RM, Freedland KE, Veith RC: Depression, the autonomic nervous system, and coronary heart disease. Psychosom Med 2005; 67:29–33Google Scholar
74. Suzuki H, Watanabe S, Hamaguchi T, et al: Brain activation associated with changes in heart rate, heart rate variability, and plasma catecholamines during rectal distention. Psychosom Med 2009; 71:619–626Google Scholar
75. Giesecke T, Gracely RH, Williams DA, et al: The relationship between depression, clinical pain, and experimental pain in a chronic pain cohort. Arthritis Rheum 2005; 52:1577–1584Google Scholar
76. Craig AD: Forebrain emotional asymmetry: a neuroanatomical basis? Trends Cogn Sci 2005; 9:566–571Google Scholar
77. Craig AD: How do you feel-now? The anterior insula and human awareness. Nature Rev (Neuroscience) 2009; 10:59–69Google Scholar
78. Rogers RC, Hermann GE: Central connections of the hepatic branch of the vagus nerve: a horseradish peroxidase histochemical study. J Auton Nerv Syst 1983; 7:165–174Google Scholar
79. Hanamori T, Kunitake T, Kato K, et al: Responses of neurons in the insular cortex to gustatory, visceral, and nociceptive stimuli in rats. J Neurophysiol 1998; 79:2535–2545Google Scholar
80. Shi CJ, Cassell MD: Cortical, thalamic, and amygdaloid connections of the anterior and posterior insular cortices. J Comp Neurol 1998; 399:440–468Google Scholar
81. Davidson RJ: What does the prefrontal cortex “do” in affect: perspectives on frontal EEG asymmetry research. Biol Psychol 2004; 67:219–233Google Scholar
82. Oppenheimer SM, Gelb A, Girvin JP, et al: Cardiovascular effects of human insular cortex stimulation. Neurology 1992; 42:1727–1732Google Scholar
83. Stephan E, Pardo JV, Faris PL, et al: Functional neuroimaging of gastric distention. J Gastrointest Surg 2003; 7:740–749Google Scholar
84. Lange CG: The mechanism of emotions, in The Classical Psychologist. Edited by Rand B. Boston, Houghton Mifflin, 1885, pp 672–685Google Scholar
85. James W: Physical basis of emotion. Psychol Rev 1894; 1:516–529. Reprinted 1994, Psychol Rev 101:205–210Google Scholar
86. Damasio AR: Descartes’ Error: Emotion, Reason and the Human Brain. New York, Grosset Putnam, 1994Google Scholar
87. Damasio AR: The Feeling of What Happens: Body and Emotion in the Making of Consciousness. New York, Harcourt Brace Jovanovich, 1999Google Scholar
88. Tye SJ, Frye MA, Lee KH: Disrupting disordered neurocircuitry: treating refractory psychiatric illness with neuromodulation. Mayo Clin Proc 2009; 84:522–532Google Scholar
89. Task Force of the European Society of Cardiology and the North American Society of Pacing and Electrophysiology: Heart rate variability: standards of measurement, physiological interpretation and clinical use. Circulation 1996; 93:1043–1065Google Scholar
90. Pagani M, Lombardi F, Guzzetti S, et al: Power spectral analysis of heart rate and arterial pressure variabilities as a marker of sympatho-vagal interaction in man and conscious dog. Circ Res 1986; 58:178–193Google Scholar

