
Vanishing White-Matter Disease: A Case of Severe Adult Onset With Prolonged Course Under Anticonvulsive Therapy
To the Editor: Leucoencephalopathy with vanishing white matter (LVWM) is a rare autosomal recessive disease, related to eIF2B subunit gene mutations.1–3 LVWM is a chronic, progressive disorder, dominated by spasticity, cerebellar ataxia, pyramidal signs, and mental impairment, with incomplete recovery and death normally within a few years.3 Typical findings include progressive white-matter rarefaction and cystic degeneration, various histopathophysiological abnormalities, and episodes of rapid and major neurologic deterioration after minor stress conditions, febrile infection, or mild head trauma.3 Although typical onset age is late infancy or early childhood, later reports have demonstrated a wider clinical spectrum of LVWM, affecting people of all ages. Milder variants, with later onset, less prominent neurologic deterioration, and protracted course have been reported.2,4,5 However, adult cases are also still rare, showing significant phenomenological variations. We present a case of an adult-onset LVWM with a prolonged course of 18 years, despite early major neurologic deterioration.
Case Report
A 36-year-old male patient was admitted to our hospital after a status epilepticus. First symptoms and epileptic seizures occurred at age 18, after a minor head injury. Until then, no neurological symptoms or cognitive decline had been reported, and the patient finished school normally. In the following months, a rapid progressive neurologic deterioration was observed, with the patient already wheelchair-bound within 1 year. Under carbamazepine 1,200 mg/day, the patient remained seizure-free and without any further neurologic deterioration for the next 7 years.
A new spontaneous onset of epileptic seizures led to the diagnosis of LVWM at the age of 25. Medical records reported “typical for LVWM gene mutation,” however, without further specification. Within the next 5 years and having ongoing seizures, the patient showed increasing spastic paraplegia, dysarthria, eye movement disorder, incontinence, cognitive decline, pathological reflexes, and was bed-bound. A change of medication to levetiracetam 1,500 mg/day and lamotrigine 500 mg/day led to another 4 seizure-free years, with no further neurologic deterioration. At age 34, another minor head injury led to a further rapid and severe neurological decline, resulting to failing verbal and nonverbal communication, major swallowing dysfunction, and new regularly emerging seizures, which lasted for the next 2 years. After admission, we increased the prescribed anticonvulsant dose to 3,000 mg/day of levetiracetam and 500 mg/day of lamotrigine, and the patient was dismissed after 2 weeks without any new seizures and under regular EEG observation.
Discussion
In our patient, the adult onset of LVWM was not accompanied by a less prominent neurologic deterioration as in other reports.2,4,5 Nevertheless, the patient showed a protracted disease course of more than 18 years. Although seizures are not the most prominent feature of the disease, we observed a major neurologic decline in our patient especially in years with frequent seizure occurrence. Seizure-free periods did not show any prominent neurological decline. We suggest that in addition to a specific phenotype and genotype, an optimal anticonvulsant treatment preventing seizures to the most possible extent could additionally contribute to a milder and prolonged course of the disease.
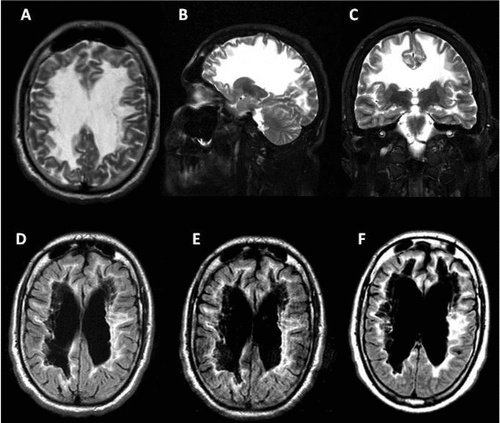
[A], [B], and [C]: Axial, sagittal and coronal brain T2-weighted image at age 30 with hemispheric white matter appearing homogeneously hyperintense.
[D], [E], and [F]: Axial FLAIR images at ages 30, 34, and 36 years, respectively, with central areas resembling signal intensity of cerebrospinal fluid surrounded by a rim of hyperintensity in the periventricular and subcortical regions. White-matter rarefaction and cystic degeneration are more evident in subcortical and periventricular regions. However, despite the progressive neurologic deterioration of the patient, only a mild progression can be noticed through MRI.
1 : Mutations in each of the five subunits of translation initiation factor eIF2B can cause leukoencephalopathy with vanishing white matter. Ann Neurol 2002; 51:264–270Crossref, Medline, Google Scholar
2 : Subunits of the translation initiation factor eIF2B are mutant in leukoencephalopathy with vanishing white matter. Nat Genet 2001; 29:383–388Crossref, Medline, Google Scholar
3 : Leukoencephalopathy with vanishing white matter: a review. J Neuropathol Exp Neurol 2010; 69:987–996Crossref, Medline, Google Scholar
4 : Natural history of adult-onset eIF2B-related disorders: a multi-centric survey of 16 cases. Brain 2009; 132:2161–2169Crossref, Medline, Google Scholar
5 : The effect of genotype on the natural history of eIF2B-related leukodystrophies. Neurology 2004; 62:1509–1517Crossref, Medline, Google Scholar

