
The Neurobiology and Pharmacotherapy of Alzheimer's Disease
Abstract
Alzheimer's disease (AD), the most common cause of dementia, has become a major public health concern as our population ages. In recent years, AD has attracted the attention of a wide range of biological disciplines, and substantial progress has been made in understanding the mechanisms of neurodegeneration in AD. Four different genes have now been associated with AD and are providing insights into the pathogenesis of the disease. The roles of beta-amyloid, tau, hormonal changes, inflammation, and oxidative stress in the neurodegeneration of AD are also being delineated. Based on these discoveries, rational therapeutic strategies are developing rapidly. The authors review these and other recent advances in the neurobiology and pharmacotherapy of AD.
Alzheimer's disease (AD), the most common cause of dementia,1 is a devastating illness characterized by cognitive deterioration as well as behavioral, affective, and psychotic disturbances. Over the past decade, dramatic progress has been made in understanding the neurogenetics and pathophysiology of AD. Four different genes have been associated with AD, and others are likely to be discovered. The mechanisms by which altered amyloid and tau protein metabolism, inflammation, oxidative stress, and hormonal changes may produce neuronal degeneration in AD are being identified, and rational pharmacological interventions based on these discoveries are being developed. The rapid pace of these developments is underscored by the more than 11,000 medline references for AD since 1990. In this article, we review recent advances in understanding the neurobiology of Alzheimer's disease and discuss corresponding therapeutic strategies.
NEURITIC PLAQUES AND NEUROFIBRILLARY TANGLES
In 1907, Alois Alzheimer reported the neuropathologic findings of Auguste D., a woman from Frankfurt who died at the age of 51 with progressive dementia.2 Alzheimer eloquently described a constellation of neuropathological abnormalities including amyloid plaques and neurofibrillary tangles. Although some controversy persists regarding the proper diagnosis of Auguste D.,3,4 the neuritic plaques (NPs) and neurofibrillary tangles (NFTs) described by Alzheimer have been considered the major pathological hallmarks of AD ever since.
Neuritic plaques are composed of extracellular spherical deposits of beta-amyloid protein (AΒ),5 which are intimately associated with dystrophic axons and dendrites as well as activated microglia and reactive astrocytes.6 The protein AΒ is composed of 39 to 43 amino acids and is produced under normal metabolic conditions from amyloid precursor protein (APP). The deposition of AΒ appears to be the earliest morphological change in the formation of neuritic plaques.7 Variants of AΒ, which differ only at their C-terminus, are found in extracellular and cerebrovascular plaques. This hydrophobic C-terminal sequence is a critical determinant of solubility, affecting the rate of amyloid formation. Soluble AΒ is composed primarily of a 40–amino acid protein (AΒ 40) with only a small amount of the 42 or 43 amino acid isoforms (AΒ 42/43). The AΒ 42/43 variant forms insoluble fibrils much faster than AΒ 40 in vitro.8 Furthermore, coincubation of small amounts of AΒ 42/43 with AΒ 40 results in immediate aggregation, suggesting that amyloid plaques may be seeded by trace amounts of this component. The term “diffuse plaques” refers to deposits of AΒ without the surrounding neuritic degeneration.9 It is believed that diffuse plaques contain the soluble, nonfibrillary form of AΒ and may represent an early stage of senile plaques.10
Neurofibrillary tangles consist of intraneuronal bundles of abnormal filaments, composed of a highly phosphorylated form of the microtubule-associated protein tau. They form either parallel bundles or randomly arranged paired helical filaments, occupying the perikaryotic cytoplasm and extending to the dendritic processes.11 The critical phosphorylation of tau depends on kinases that are still being identified.
The pathological features of AD occur in a consistent pattern. On a cellular level, pyramidal neurons, the main component of corticocortical pathways,12 are the major cortical cell type lost in AD.13 The majority of the NFTs are seen in cortical layers III and V, whereas neuritic plaques, present throughout the depth of the cortex, are more numerous in layers II and III.12 NPs are initially found predominantly in the amygdala and in high-order temporal and parietal association cortex. Later in the course of the disease, they develop in other mesial temporolimbic structures such as the hippocampus.14 In addition, NPs are found in meningeal and cortical blood vessels. Primary visual and sensorimotor areas remain relatively spared.12 NFTs have a slightly different distribution. Although NFTs are abundant in high-order association cortex, the entorhinal cortex, hippocampus, and amygdala are affected early in the disease process.15 NFTs are not specific for AD and may develop in response to various cerebral insults such as postencephalitic parkinsonism, subacute sclerosing panencephalitis, and dementia pugilistica, among other conditions. Both NFTs and NPs are seen in normal aging, although their density and distribution typically differ from that seen in AD. Current neuropathologic criteria for the diagnosis of AD are based on the quantitative distribution of NPs.16,17
APP METABOLISM
Amyloid precursor protein is an integral membrane glycoprotein (see review6). Within APP, AΒ encompasses 28 residues of the extracellular and 12 to 15 residues of the putative transmembrane domains. APP occurs in different isoforms, the 695–amino acid form being expressed almost exclusively in neurons. The two other common forms (751 and 770 amino acids) are expressed in both neural and non-neural cells and carry an additional domain with protease inhibitor functions. Transported to the cell membrane via secretory vesicles, some of these molecules undergo proteolytic cleavage by an unidentified protease, termed α-secretase. This process releases a soluble ectodomain, designated “α APP.” It also cleaves the precursor within the AΒ domain, precluding AΒ protein formation.18 The basal release of the soluble form of APP appears to be regulated in vitro by receptor-coupled activation of protein kinase C.19,20 Alternatively, uncleaved surface APP may follow a proteolytic pathway. APP is internalized and recycled by endosome-lysosomes21,22 or via an endocytic route.23 This latter pathway seems to be the site of subsequent cleavage by the putative β- and γ-secretases, which results in production and release of AΒ.23 Increased intracellular calcium levels enhance AΒ production, suggesting that calcium-requiring protease(s) may be involved in the production of AΒ.24
APP GENE AND CHROMOSOME 21
The observation that patients with Down's syndrome (trisomy 21) develop cognitive deterioration and typical pathological features of AD by middle age led to the discovery of the APP gene on chromosome 21.25 Simultaneously, a locus segregating with a minority of early-onset familial AD (FAD) kindreds was mapped to this chromosome26 in the same region as the APP gene.27 Later, several missense mutations within the APP gene that resulted in amino acid substitutions in APP were identified in these FAD kindreds.28–32 It appears that such mutations alter the previously described proteolytic processing of APP, generating amyloidogenic forms of AΒ. Skin fibroblasts from subjects carrying APP mutations produce increased AΒ 42/43.33 Increased plasma concentration of AΒ 42/43 is also seen in these subjects34 regardless of age, sex, or clinical status.33,34 Interestingly, some patients with sporadic AD may exhibit similar elevations of plasma AΒ 42/43.34
Improved understanding of amyloid metabolism provides several theoretical targets for pharmacological interventions. Since the basal release of the soluble form of APP is dependent on putative α-secretase and regulated by protein kinase C activity, agents that enhance this metabolic pathway may directly impede AΒ formation. Conversely, drugs that decrease cleavage of APP by β- and γ-secretases might also decrease AΒ deposition. In addition, agents that inhibit the aggregation of AΒ may diminish its neurotoxicity. Transgenic mice expressing human mutant APP have recently been developed. They will likely prove useful in assessing these and other potential therapeutic agents. These mice develop memory deficits35,36 and exhibit many of the pathologic abnormalities seen in AD, including AΒ deposits, neuritic plaques, synapse loss, astrocytosis and activated microglial cells.37 They do not produce NFTs.
PRESENILIN-1 AND PRESENILIN-2
Fifty to seventy percent of early-onset autosomal dominant AD cases appear to be associated with a locus (AD3) mapped by genetic linkage to the long arm of chromosome 14 (14q24.3).38 Numerous missense mutations have been identified on a strong candidate gene called presenilin-1 (PS1).39,40 At the same time, another autosomal dominant locus responsible for early-onset AD was localized to chromosome 1. Two mutations were identified on the candidate gene, designated presenilin-2 (PS2). One was demonstrated in an Italian pedigree,41 and the other one was found in Volga German kindreds.42 The physiological role of presenilins and the pathogenic effects of their mutations are not yet well understood. Their two putative products, PS1 and PS2, share substantial amino-acid and structural similarities, suggesting that they may be functionally related. In addition, the expression patterns of PS1 and PS2 in the brain are very similar, if not identical. Both PS1 mRNA and PS2 mRNA are detectable only within neuronal populations.43 Mutations result in single amino-acid substitutions in PS1 and PS2.43 PS1 and PS2 exhibit strong homology with two peptides of a nematode (Caenorhabditis elegans) involved in the intracellular trafficking and recycling of proteins. Immunochemical analyses indicate that PS1 localize to intracellular compartments such as the endoplasmic reticulum and Golgi complex43,44 that are involved in similar functions. Recent evidence supports the role of presenilins in AΒ metabolism. Mice deficient in the expression of PS1 exhibit dramatic decrease in proteolytic cleavage by γ-secretase of the transmembrane domain of APP.45
Presenilin-1 is immunoreactive with NPs.46,47 Asymptomatic and demented subjects carrying the PS1 mutation have increased production of the amyloidogenic AΒ 42/43 isoform in skin fibroblasts and plasma.34 Prominent deposition of AΒ 42/43 is found in many brain regions of patients with PS1 mutations.48 These findings, in suggesting that inhibiting presenilin function might decrease AΒ amyloid production, offer new therapeutic avenues.
APOLIPOPROTEIN ALLELES AND CHROMOSOME 19
The gene encoding the cholesterol-carrying apolipoprotein E (APOE) on chromosome 19 has been linked to late-onset familial49 and sporadic50 AD. This gene is inherited as an autosomal codominant trait with three alleles. APOE E3 is the most frequent allele (allele frequency=0.78), followed by APOE E4 (allele frequency=0.14) and APOE E2 (allele frequency=0.07). Three homozygous genotypes (APOE E4/E4, E3/E3, and E2/E2) and three heterozygous genotypes (APOE E3/E4, E3/E2, and E2/E4) result in the respective phenotypes.51 APOE E4 gene “dose” is correlated with increased risk49,52 and earlier onset of AD. Subjects with 2 copies of the APOE E4 allele (4/4 genotype) have significantly greater risk of developing AD than subjects with 2/3 or 3/3 genotypes. On the other hand, the APOE E2 allele may confer a protective effect.53,54 Mean age at onset is significantly lower in the presence of two APOE E4 copies.49,54 A recent collaborative study suggested that APOE E4 exerts its maximal effect before the age of 70.55 Many APOE E4 carriers do not develop AD,56 and many patients with AD do not have this allele. Therefore, the presence of an APOE E4 allele does not secure the diagnosis of AD, but instead the APOE E4 allele acts as a biological risk factor for the disease,52,56 especially before the age of 70.
How APOE genotype influences AD susceptibility remains unclear. Subjects with one or two copies of APOE E4 allele exhibit more beta amyloid staining at autopsy than subjects homozygous for APOE E3,57 suggesting that APOE genotype may affect the rate of amyloidogenesis. APOE is synthesized in various organs including the brain.51 The APOE E4 isoform binds to AΒ with higher affinity than the other APOE E isoforms.58,59 APOE is also immunoreactive with plaques in situ.57 Studies have suggested that the pathogenic role of APOE E4 may be the result of inefficient suppression of amyloidogenesis60 or increased AΒ polymerization.61 Impaired clearance of AΒ with sequestration in the extracellular space50 has also been suggested.
However, APOE may be involved in microtubular metabolism as well. APOE is immunoreactive with neurofibrillary tangles in situ,62 and electron microscopic studies have localized APOE to the cytoplasm (see review63). There are also APOE-isoform differences in binding to tau with higher avidity for APOE E3 than for APOE E4.62 APOE E3 binds to the receptor-binding domain of tau thought to be involved in the assembly of paired helical filaments.63 There are also APOE-isoform differences in neurite outgrowth. APOE E4 is associated with depolymerization of microtubules and inhibition of neurite outgrowth.64 By contrast, APOE E3 is believed to prevent the hyperphosphorylation of tau and thereby to protect tau's abilities to interact with cellular microtubules, stabilize the cytoskeleton, and support neurite elongation.65
THE AMYLOID HYPOTHESIS VERSUS THE TAU HYPOTHESIS
A central and controversial issue in the pathogenesis of AD is the relationship between amyloid deposition and NFT formation. That abnormal amyloid metabolism plays a key pathogenic role is supported by several lines of evidence. The fibrillar form of AΒ has been shown to be neurotoxic to cultured neurons at high concentrations.66,67 Apoptosis, self-regulated cell destruction, is one possible mechanism of cellular death in AD.68 Cultured cortical and hippocampal neurons treated with AΒ protein exhibit changes characteristic of apoptosis, including nuclear chromatin condensation, plasma membrane blebbing, and internucleosomal DNA fragmentation.68 The fibrillar form of AΒ has also shown to alter the phosphorylation state of tau protein.69 The identification of several point mutations within the APP gene in some patients with early-onset familial AD and the development of transgenic mice exhibiting cognitive changes and NPs also incriminate AΒ in AD. Additional evidence comes from recent experimental data supporting the role of presenilins in AΒ metabolism as well as findings of abnormal production of AΒ protein in presenilin-mutation familial Alzheimer's disease.
Although very popular, the “amyloid hypothesis” is not uniformly accepted. Dementia severity correlates better with the number of neocortical NFTs than with NPs.15 Tau is a protein whose function is to stabilize neuronal microtubules.65 As mentioned previously, the APOE E3 allele may protect tau from hyperphosphorylation. It has been speculated that destabilization of the microtubular system may disrupt the Golgi apparatus, in turn inducing abnormal protein processing and increasing production of AΒ.70 In addition, this destabilization may decrease axoplasmic flow, generating dystrophic neurites and contributing to synaptic loss.
OXIDATIVE STRESS
Oxidative stress is believed to be a critical factor in normal aging71 and neurodegenerative diseases such as Parkinson's disease and amyotrophic lateral sclerosis. There is evidence that oxidative stress is also implicated in AD. Formation of free carbonyls72 and thiobarbituric acid-reactive products,73 an index of oxidative damage, are significantly increased in AD brain tissue compared with age-matched controls. Plaques and tangles display immunoreactivity to antioxidant enzymes.74 There are multiple mechanisms by which cellular alterations may be induced by oxidative stress, including production of reactive oxygen species (ROS) in the cell membrane (lipid peroxidation). This in turn impairs the various membrane proteins involved in ion homeostasis75,76 such as N-methyl-d-aspartate receptor channels or ion-motive adenosine triphosphatases.75 The subsequent increase in intracellular calcium, along with the accumulation of ROS, damages various cellular components such as proteins, DNA, and lipids and may result in apoptotic cellular death.75,76 Increased intracellular calcium may also alter calcium-dependent enzyme activity77 such as the implication of protein kinase C in amyloid protein metabolism and the phosphorylation of tau. Therefore, blocking the increase in free intracellular calcium may diminish neuronal injury. Nimodipine is a lipophilic calcium channel blocker whose activity is mediated through inactivation of voltage-dependent L-type (long-lasting) calcium channels. Clinical trials of nimodipine for AD have yielded generally disappointing results.78
The apoptotic pattern of cellular death seen in oxidative stress is similar to that produced by AΒ exposure,68 and AΒ neurotoxicity is attenuated by antioxidants such as vitamin E.75 AΒ may induce its toxicity by engaging several binding sites on the membrane surface.66 The receptor for advanced glycation end products (RAGE) may be one of these receptors.79 RAGE is a member of the immunoglobulin superfamily of cell surface molecules known for its capacity to bind advanced glycation end products. This receptor is highly expressed by cortical neurons, especially in the hippocampus and cerebellum, during rat brain development.80 RAGE is also expressed in a variety of other cell types, including endothelial cells and mononuclear phagocytes. Activation of this receptor is believed to trigger cellular oxidative reactions. In addition, RAGE has been shown to mediate the interaction of AΒ protein with glial cells, which may be one of the first steps in the inflammatory cascade (see below).
The long-term effects of alpha-tocopherol (vitamin E) and l-deprenyl (selegiline) on AD progression were recently assessed.81 Vitamin E has antioxidant properties, and selegiline, an irreversible inhibitor of monoamine oxidase B (MAO-B), is believed to act as free radical scavenger and modulator of monoaminergic neurotransmission. Selegiline is marketed for the treatment of Parkinson's disease. In a controlled trial, 341 patients with AD were treated with selegiline (10 mg/day), vitamin E (2,000 IU/ day), both, or placebo over a 2-year period.81 After adjustment for baseline differences among the groups on the Mini-Mental State Examination, both vitamin E and selegiline were found to delay the time of occurrence of death, institutionalization, and loss of the ability to perform activities of daily living. However, there was no significant difference between the different groups on the cognitive section of the Alzheimer's Disease Assessment Scale (ADAS-Cog), a 70-point cognitive battery designed to provide a global index of cognitive performances.82 A series of short-term studies of selegiline has suggested a beneficial effect in AD (reviewed83), but long-term studies have showed no or very mild improvement on cognitive measures.84–86 Milacemide and lazabemide are two other MAO-B inhibitors. Milacemide has not proven to be effective in AD.87 Lazabemide is now under investigation in the United States. A study from Bordeaux investigated the relationship between alcohol consumption and risk of AD. A negative association was found between “moderate” alcohol consumption (3 to 4 glasses of red wine per day) and the incidence of AD.88 One hypothesized explanation rests on the antioxidant properties of red wine.89 The effects of ginkgo biloba, a compound with putative antioxidant properties, were recently evaluated in a study of 236 AD patients.90 A modest but statistically significant improvement was seen on the ADAS-Cog in the ginkgo-treated group. Idebenone is an antioxidant that inhibits lipid peroxidation in vitro. This compound is currently in phase III clinical trials in the United States. Recent experimental data have suggested that melatonin may have neuroprotective effects by preventing AΒ-induced oxidative damage.91 Melatonin prevented increased intracellular calcium, lipid peroxidation, and apoptotic death in neuroblastoma cells exposed to AΒ. It is interesting to note that serum92 and cerebrospinal fluid93 melatonin levels are reduced in demented subjects compared with elderly control subjects. Further trials are needed to delineate the clinical application of this compound.
INFLAMMATORY REACTIONS
A growing body of evidence indicates that inflammatory and immune mechanisms accompany the degenerative process in AD. Reactive microglia, cells phenotypically related to peripheral monocytes, are densely embedded in neuritic plaques.94–96 Increased cytokines are seen in the serum, cortical plaques, and neurons of patients with AD compared with aged-matched control subjects.97 Interestingly, the anti-inflammatory cytokine-transforming growth factor beta 1 (TGF-β1) has recently been found to promote or accelerate the deposition of AΒ.98 Classical complement pathway fragments are also found in the brains of AD patients, and AΒ may directly activate the classical complement pathway in an antibody-independent fashion.96 One recent study found an association between AD and the major histocompatibility complex, located on chromosome 6.99 The age at onset of AD was significantly lower in patients carrying the A2 allele. The possibility of linkage disequilibrium with another gene requires further study.
The potential role of anti-inflammatory agents in the treatment of AD was first suggested by the discovery of an unexpectedly low prevalence of AD among patients with rheumatoid arthritis.94,100 Additional case-control,101 cross-sectional,102 and longitudinal studies103 have found an inverse relation between the risk for AD and nonsteroidal anti-inflammatory drug (NSAID) use. The relative risk for AD decreased with duration of NSAID use, from 0.65 for a less than 2-year exposure to 0.40 for 2 or more years of treatment.103 The protection conferred by aspirin appears weaker than that of NSAIDs, and no association was found between AD risk and use of acetaminophen. A 1-year longitudinal study found that AD patients using NSAIDs declined less on several cognitive measures.104 A 6-month double-blind placebo-controlled trial of indomethacin has been conducted in 44 patients suffering from mild to moderate AD. Given at a dose of 100–150 mg/day, indomethacin was found to improve cognitive function.105 However, about 20% of patients developed drug-related side effects, primarily gastrointestinal intolerance. The toxicity of long-term NSAID use, especially in elderly patients, may hinder the potential use of these agents in patients with, or at risk for, AD. Recent observations suggest that inhibition of cyclo-oxygenase (COX)-1 may cause much of this toxicity, whereas inhibition of COX-2 may confer the clinical anti-inflammatory effect. Interestingly, COX-2 is expressed in neocortex, particularly in limbic structures such as the hippocampus that are preferentially involved in Alzheimer's disease.106 Selective inhibitors of COX-2 are currently under investigation for the treatment of AD.
Whether markers of immune and inflammatory processes actively participate in the neurodegenerative process or whether they represent an epiphenomenon remains unclear. Brain specimens from elderly patients with arthritis treated with NSAIDs have similar numbers of senile plaques, as do control brains. However, less microglial activation is seen in the arthritis patients' brains.107 This suggests that although NSAIDs may not impede senile plaque formation, they may delay or prevent clinical symptoms by limiting the associated inflammation.
As mentioned above, RAGE has been shown to mediate the interaction of AΒ and glial cells, producing cellular activation and an inflammatory response with cytokine production, chemotaxis, and haptotaxis. The expression of this receptor appears to be upregulated in neurons, vasculature, and microglia in affected regions of AD brains.79 The unrelated class A scavenger receptor (Class A SR) also mediates the adhesion of microglial cells to AΒ fibrils. Senile plaques contain high concentrations of microglia that express class A SRs.108 RAGE and class A SRs may represent novel pharmacological targets for diminishing the inflammatory and oxidative reactions associated with AD.
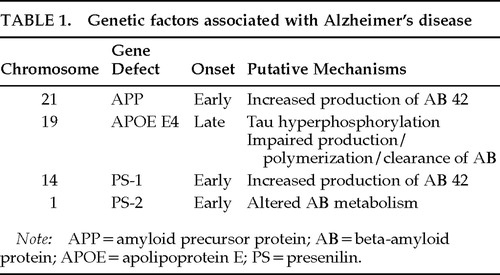
Genetic factors associated or potentially associated with Alzheimer's disease are summarized in Table 1 and Table 2.
ESTROGENS AND ESTROGEN REPLACEMENT THERAPY
Experimental studies have demonstrated that estrogen loss predisposes to cognitive decline and neuronal degeneration (see review112). For instance, ovariectomized female rats have diminished learning abilities. Neuronal choline uptake and choline acetyltransferase activities, indices of cholinergic function, are shown to be reduced in the brain of these animals. The expression of nerve growth factor and brain-derived neurotrophic factor mRNA is decreased as well. These cognitive and biological changes are reduced by administration of estrogen replacement therapy (ERT). In addition, estrogen has been shown to exert cytoprotective effects and to prevent AΒ toxicity in human neuroblastoma cell cultures.
Human studies suggest that ERT may prevent, delay, or reduce the symptoms of AD. In a case control study, treatment with ERT at the time of enrollment was found to be significantly less common in women meeting criteria for AD than in control subjects.113 In addition, the demented women using ERT performed significantly better on the Mini-Mental State Examination. Confounding factors included retrospective analysis and the lack of information regarding dose and previous history of ERT. Another study, with a similar design, found comparable results.114 A beneficial role for postmenopausal ERT is more convincingly supported by longitudinal studies of normal aging. In a group of 1,124 elderly women,115 age at onset of AD was significantly later among estrogen users. The relative risk of AD was significantly reduced even after adjustment for education, ethnic origin, and apolipoprotein E genotype. This was especially true for women on ERT for longer than 1 year. Another study of 472 post- and perimenopausal women provided additional support for a protective effect of ERT.116 The observational design of these two studies resulted in several limitations involving nonrepresentative populations and variability in dose and route of administration. Prospective trials of estrogen in normal perimenopausal women are in progress.
Little information is available on the effect of ERT in AD patients. In a double-blind placebo-controlled multicenter clinical trial of tacrine for AD, a subgroup of women receiving ERT had a significantly better response than women receiving tacrine alone or placebo.117 A randomized clinical trial of ERT in women with AD is currently in progress (Alzheimer's Disease Cooperative Study).
CHOLINERGIC NEUROTRANSMISSION AND ALZHEIMER'S DISEASE
Cholinergic systems are involved in normal memory functions,118 and cholinergic deficiency has been implicated in the cognitive and behavioral119 manifestations of AD. Activity of the synthetic enzyme choline acetyltransferase (CAT) and the catabolic enzyme acetylcholinesterase (ACE) are significantly reduced in the cerebral cortex,120,121 hippocampus, and amygdala in AD patients.122 The nucleus basalis of Meynert, medial septum, and diagonal band of Broca, which provide the main cholinergic input to the hippocampus, amygdala, and neocortex, demonstrate a fairly selective pattern of neuronal degeneration in AD.123 Loss of cortical CAT124 and decline in acetylcholine synthesis in biopsy specimens have been found to correlate with cognitive impairment125 and reaction time performance.126 The relationship between basal forebrain dysfunction and amyloid precursor protein metabolism remains poorly understood. Experimental lesions of the nucleus basalis in rats result in elevated cerebrospinal fluid APP secretion.127 The effects of cholinergic agents on APP release have yielded conflicting results in animal studies.
Because cholinergic dysfunction may contribute to the symptoms of AD patients,118 enhancing cholinergic neurotransmission constitutes a rational basis for symptomatic treatment. Attempts to augment cholinergic activity have employed acetylcholine (ACh) precursors, cholinergic agonists, and drugs inhibiting acetylcholinesterase (AChE), the enzyme responsible for the breakdown of ACh. Precursor therapy, using choline or phosphatidylcholine (lecithin), has been largely unsuccessful.128 Thus far, cholinergic agonists (bethanechol, pilocarpine, arecoline, RS-86, xanomeline tartrate) have yielded modest results or have been associated with significant side effects.129,130
Various classes of AChE inhibitors have been studied for the treatment of AD. They differ in their mechanism of inhibition (reversible, pseudo-irreversible, or irreversible), metabolism, and brain selectivity. Physostigmine, a short-acting cholinesterase inhibitor, was one of the first drugs offering promising results. In the initial study, intravenous physostigmine enhanced performances on a recognition memory task.130 A review of 11 studies using physostigmine131 found improvement in all five investigations that involved parenteral administration and in four of the six involving oral administration. Tacrine (THA or tetrahydroaminoacridine) is a centrally active reversible AChE inhibitor with a moderately long duration of action. It is thought to exhibit a variety of other pharmacological properties such as enhancement of monoaminergic activity. In 1986, Summers et al.132 reported significant improvement in the performance of psychometric tests after long-term treatment with tacrine. Larger multicenter studies confirmed these findings, with statistically significantly improvement on various outcome measures such as the ADAS-Cog,133–135 clinician's impression scales,134,135 and time until nursing home placement.136 Elevation of serum levels of alanine aminotransferase (ALT), greater than three times the normal value, occurs in about 25% of patients.137 Adverse events also included gastrointestinal complaints, and resulted in frequent dosage adjustment and a high dropout rate.135–137 Overall, these studies demonstrated that tacrine produced modest, dose-dependent, symptomatic improvement in a portion of patients who were able to tolerate it.
Donepezil inaugurates a new generation of AChE inhibitors with greater selectivity for AChE in the central nervous system. Donepezil is a reversible AChE inhibitor with a long plasma half-life of 70 hours. A recent double-blind study enrolled 161 patients with mild to moderate AD for a period of 15 weeks.138 Subjects received either placebo or daily doses of 1 mg, 3 mg, or 5 mg of donepezil. There was statistically significant, dose-related improvement in ADAS-Cog scores in the donepezil group. Another 30-week study of 473 patients yielded similar results. After 6 months, the mean improvement in the ADAS-Cog scores for the donepezil group was 3 points compared with the placebo group.139 Donepezil has not been associated with hepatotoxicity. Nausea, vomiting, and diarrhea led to discontinuation in less than 3 percent of patients.
Tacrine and donepezil have been approved by the Food and Drug Administration (FDA) for the treatment of mild to moderate AD. For tacrine, initial FDA-approved dosing strategy recommended starting at 10 mg four times daily and increasing by 40 mg every 6 weeks to a maximum of 160 mg. Transaminase levels were monitored every other week, and tacrine was to be stopped if ALT levels exceeded 5 times their normal value. New recommendations are expected. They should allow increasing the doses every 4 weeks, and include a monitoring of transaminases every 4 weeks, to be discontinued if ALT levels exceed 20 times their normal value. For donepezil, dosing recommendations suggest starting the treatment with 5 mg daily. The possibility of additional benefits with a higher dose allows increases up to 10 mg after 4 to 6 weeks.
Although still under investigation, rivastigmine (ENA 713), a carbamate AChE inhibitor, and metrifonate, an organophosphorus compound used as an insecticide and for the treatment of schistosomiasis, produce no liver toxicity and may hold promise in the treatment of AD.
The locus ceruleus provides the principal noradrenergic innervation of cerebral cortex, whereas the dorsal raphe nucleus and central superior nucleus provide serotonergic projections. In AD, neuronal loss and NFTs are seen in both these structures.140,141 Some studies have shown an association between locus ceruleus or central superior nucleus involvement and depression in patients with AD.141,142 A variety of other neurotransmitter and neuropeptide systems are also abnormal in AD, including dopamine, somatostatin, corticotrophin-releasing factor, GABA, and substance P, suggesting that repleting any one neurotransmitter is unlikely to produce dramatic clinical improvement.
CONCLUSION
Alzheimer's disease results from multiple pathogenic events, including protein processing abnormalities, inflammation, and oxidative stress, leading to the destruction of selective neuronal populations. Figure 1 diagrams the main pathologic events and potential pharmacological targets in AD. Until recently, no effective treatments were available for patients with Alzheimer's disease. Cholinergic enhancement with acetylcholinesterase inhibitors has now been conclusively shown to provide mild symptomatic benefit in some patients with AD. Estrogen replacement therapy may provide additional symptomatic benefit and may slow disease progression. Anti-inflammatory and antioxidant medications may also directly interfere with the underlying disease process. Improved understanding of amyloid metabolism offers a theoretical framework for multiple pharmacological interventions. Ultimately, the optimal pharmacological management of AD will likely consist of polypharmacy with agents that enhance deficient neurotransmitters and target multiple steps of the underlying disease process.
 |
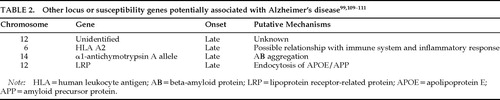 |
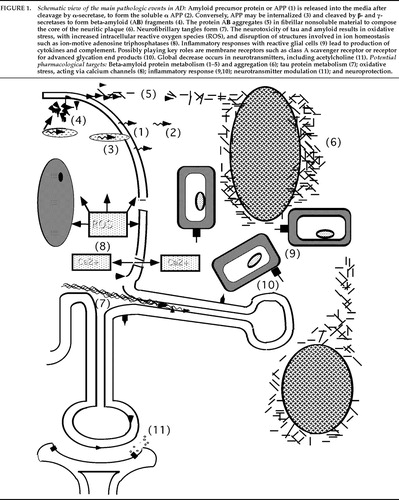
FIGURE 1. Schematic view of the main pathologic events in AD: Amyloid precursor protein or APP (1) is released into the media after cleavage by α-secretase, to form the soluble α APP (2). Conversely, APP may be internalized (3) and cleaved by β- and γ-secretases to form beta-amyloid (AΒ) fragments (4). The protein AΒ aggregates (5) in fibrillar nonsoluble material to compose the core of the neuritic plaque (6). Neurofibrillary tangles form (7). The neurotoxicity of tau and amyloid results in oxidative stress, with increased intracellular reactive oxygen species (ROS), and disruption of structures involved in ion homeostasis such as ion-motive adenosine triphosphatases (8). Inflammatory responses with reactive glial cells (9) lead to production of cytokines and complement. Possibly playing key roles are membrane receptors such as class A scavenger receptor or receptor for advanced glycation end products (10). Global decrease occurs in neurotransmitters, including acetylcholine (11). Potential pharmacological targets: Beta-amyloid protein metabolism (1–5) and aggregation (6); tau protein metabolism (7); oxidative stress, acting via calcium channels (8); inflammatory response (9,10); neurotransmitter modulation (11); and neuroprotection.
1 Tomlinson BE, Blessed G, Roth M: Observations on the brains of demented people. J Neurol Sci 1970; 11:205–242Crossref, Medline, Google Scholar
2 Alzheimer A: Uber eine eigenartige Erkrankung der Hirnrinde. Zeitschrift für Psychiatrie 1906; 10:146–148Google Scholar
3 Maurer K, Volk S, Gerbaldo H: Auguste D and Alzheimer's disease. Lancet 1997; 349:1546–1549Crossref, Medline, Google Scholar
4 Amaducci L: Alzheimer's original patient (letter). Science 1996; 274:328Crossref, Medline, Google Scholar
5 Glenner GG, Wong CW: Alzheimer's disease: report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun 1984; 120:885–890Crossref, Medline, Google Scholar
6 Selkoe DJ: Amyloid beta-protein and the genetics of Alzheimer's disease. J Biol Chem 1996; 271:18295–18298Crossref, Medline, Google Scholar
7 Mann DMA, Esiri MM: The pattern of acquisition of plaques and tangles in the brain of patients under 50 years of age with Down's syndrome. J Neurol Sci 1989; 89:169–179Crossref, Medline, Google Scholar
8 Jarret JT, Berger EP, Lansbury PT: The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer's disease. Biochemistry 1993; 32:4693–4697Crossref, Medline, Google Scholar
9 Dekosky ST: Advances in the biology of Alzheimer's disease, in The Dementias: Diagnosis, Management, and Research, 2nd edition, edited by Weiner MF. Washington, DC, American Psychiatric Press, 1996, pp 313–330Google Scholar
10 Bugiani O, Tagliavini F, Giaccone G, et al: Diffuse senile plaques: amorphous or fibrous. Am J Pathol 1995; 146:777–778Medline, Google Scholar
11 Ohtsubo K, Izumiyama N, Shimada H, et al: Three-dimensional structure of Alzheimer's neurofibrillary tangles of the aged human brain revealed by the quick-freeze, deep-etch and replica method. Acta Neuropathol 1990; 79:480–485Crossref, Medline, Google Scholar
12 Pearson RCA, Esiri MM, Hiorns RW, et al: Anatomical correlates of the distribution of the pathological changes in the neocortex in Alzheimer disease. Proc Natl Acad Sci USA 1985; 82:4531–4534Crossref, Medline, Google Scholar
13 Terry RD, Peck A, DeTeresa R, et al: Some morphometric aspects of the brain in senile dementia of the Alzheimer type. Ann Neurol 1981; 10:184–192Crossref, Medline, Google Scholar
14 Giannakopoulos P, Hof PR, Giannakopoulos A, et al: Regional distribution of neurofibrillary tangles and senile plaques in the cerebral cortex of very old patients. Arch Neurol 1995; 52:1150–1159Crossref, Medline, Google Scholar
15 Arriagada PV, Growdon JH, Hedley-Whyte T, et al: Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology 1992; 42:631–639Crossref, Medline, Google Scholar
16 Khachaturian ZS: Diagnosis of Alzheimer's disease. Arch Neurol 1985; 42:1097–1104Crossref, Medline, Google Scholar
17 Mirra SS, Heyman A, McKeel D, et al: The Consortium to Establish a Registry for Alzheimer's Disease (CERAD), II: standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 1991; 41:479–486Crossref, Medline, Google Scholar
18 Esch FS, Keim PS, Beattie EC, et al: Cleavage of amyloid beta peptide during constitutive processing of its precursor. Science 1990; 248:1122–1124Crossref, Medline, Google Scholar
19 Nitsch RN, Slack BE, Wurtman RJ, et al: Release of Alzheimer amyloid. Science 1992; 258:304–307Crossref, Medline, Google Scholar
20 Caporaso GL, Gandy SE, Buxbaum JD, et al: Protein phosphorylation regulates secretion of Alzheimer beta A4 amyloid precursor protein. Proc Natl Acad Sci USA 1992; 89:3055–3059Crossref, Medline, Google Scholar
21 Haass C, Koo EH, Mellon A, et al: Targeting of cell-surface beta-amyloid precursor protein to lysosomes: alternative processing into amyloid-bearing fragments. Nature 1992; 357:500–503Crossref, Medline, Google Scholar
22 Golde TE, Estus S, Younkin LH, et al: Processing of the amyloid protein precursor to potentially amyloidogenic derivatives. Science 1992; 255:728–730Crossref, Medline, Google Scholar
23 Koo EH, Squazzo SL: Evidence that production and release of amyloid beta-protein involves the endocytic pathway. J Biol Chem 1994; 269:17386–17389Medline, Google Scholar
24 Querfurth HW, Selkoe DJ: Calcium ionophore increases amyloid beta peptide production by cultured cells. Biochemistry 1994; 33:4550–4561Crossref, Medline, Google Scholar
25 Goldgaber D, Lerman MI, McBride OW, et al: Characterization and chromosomal localization of a cDNA encoding brain amyloid of Alzheimer's disease. Science 1987; 235:877–880Crossref, Medline, Google Scholar
26 St George Hislop PH, Tanzi RE, Polinsky RJ, et al: The genetic defect causing familial Alzheimer's disease maps on chromosome 21. Science 1987; 235:885–890Crossref, Medline, Google Scholar
27 Patterson D, Gardiner K, Kao FT, et al: Mapping of the gene encoding the beta-amyloid precursor protein and its relationship to the Down syndrome region of chromosome 21. Proc Natl Acad Sci USA 1988; 85:8266–8270Crossref, Medline, Google Scholar
28 Goate A, Chartier-Harlin MC, Mulan M, et al: Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature 1991; 349:704–706Crossref, Medline, Google Scholar
29 Murrell J, Farlow M, Ghetti B, et al: A mutation in the amyloid precursor protein associated with hereditary Alzheimer's disease. Science 1991; 254:97–99Crossref, Medline, Google Scholar
30 Chartier-Harlin M, Crawford F, Houlden H, et al: Early onset Alzheimer's disease caused by mutations at codon 717 of the beta-amyloid precursor protein gene. Nature 1991; 353:844–846Crossref, Medline, Google Scholar
31 Mullan M, Houlden H, Windelspecht M, et al: A locus for familial early onset Alzheimer's disease on the long arm of chromosome 14, proximal to the alpha 1-antichymotrypsin gene. Nat Genet 1992; 2:340–342Crossref, Medline, Google Scholar
32 Hendriks L, Van Dujn CM, Cras P, et al: Presenile dementia and cerebral haemorrhage linked to a mutation at codon 692 of the beta-amyloid precursor protein gene. Nat Genet 1992; 1:218–221Crossref, Medline, Google Scholar
33 Citron M, Vigo-Pelfrey C, Teplow DB, et al: Excessive production of amyloid beta-protein by peripheral cells of symptomatic and presymptomatic patients carrying the Swedish familial Alzheimer's disease mutation. Proc Natl Acad Sci USA 1994; 91:11993–11997Crossref, Medline, Google Scholar
34 Scheuner D, Eckman C, Jensen M, et al: Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat Med 1996; 2:864–870Crossref, Medline, Google Scholar
35 Hsiao K, Chapman P, Nilsen S, et al: Correlative memory deficits, A-beta elevation, and amyloid plaques in transgenic mice. Science 1996; 274:99–102Crossref, Medline, Google Scholar
36 Nalbantoglu J, Tirado-Santiago G, Lahsaini A, et al: Impaired learning and LTP in mice expressing the carboxy terminus of the Alzheimer amyloid precursor protein. Nature 1997; 387:500–505Crossref, Medline, Google Scholar
37 Games D, Adams D, Alessandrini R, et al: Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature 1995; 373:523–527Crossref, Medline, Google Scholar
38 Shellenberg GD, Bird TD, Wijsman EM, et al: Genetic linkage evidence for a familial Alzheimer's disease locus on chromosome 14. Science 1992; 258:668–671Crossref, Medline, Google Scholar
39 Sherrington R, Rogaev EI, Liang Y, et al: Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature 1995; 375:754–760Crossref, Medline, Google Scholar
40 Sandbrink R, Zhang D, Schaeffer S, et al: Missense mutations of the PS-1/S182 gene in German early-onset Alzheimer's disease patients. Ann Neurol 1996; 2:265–266Crossref, Google Scholar
41 Rogaev EI, Sherrington R, Rogaeva EA, et al: Familial Alzheimer disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer disease type 3 gene. Nature 1995; 376:775–778Crossref, Medline, Google Scholar
42 Levy-Lahad E, Wasco W, Poorkaj P, et al: Candidate gene for the chromosome 1 familial Alzheimer's disease locus. Science 1995; 269:973–977Crossref, Medline, Google Scholar
43 Kovacs DM, Fausett HJ, Page KJ, et al: Alzheimer-associated presenilins 1 and 2: neuronal expression in brain and localization to intracellular membranes in mammalian cells. Nat Med 1996; 2:864–870Crossref, Medline, Google Scholar
44 Cook DG, Sung JC, Golde TE, et al: Expression and analysis of presenilin 1 in a human neuronal system: localization in cell bodies and dendrites. Proc Natl Acad Sci USA 1996; 93:9223–9228Crossref, Medline, Google Scholar
45 De Strooper B, Saftig P, Craessaerts K, et al: Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 1998; 391:387–390Crossref, Medline, Google Scholar
46 Wisniewski T, Palha JA, Ghiso J, et al: S182 protein in Alzheimer's disease neuritic plaques. Lancet 1995; 346:1366Crossref, Medline, Google Scholar
47 Giannakopoulos P, Bouras C, Kovari E, et al: Presenilin-1-immunoreactive neurons are preserved in late-onset Alzheimer's disease. Am J Pathol 1997; 150:429–436Medline, Google Scholar
48 Lemere CA, Lopera F, Kosik KS, et al: The E280A presenilin 1 Alzheimer mutation produces increased Abeta 42 deposition and severe cerebellar pathology. Nat Med 1996; 2:1146–1150Crossref, Medline, Google Scholar
49 Corder EH, Saunders AM, Strittmatter WJ, et al: Gene dose of Apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 1993; 261:921–923Crossref, Medline, Google Scholar
50 Saunders AM, Strittmatter WJ, Schmechel D, et al: Association of apolipoprotein E allele epsilon4 with late-onset familial and sporadic Alzheimer's disease. Neurology 1993; 43:1467–1472Crossref, Medline, Google Scholar
51 Mahley RW: Apolipoprotein E: cholesterol transport protein with expending role in cell biology. Science 1988; 240:622–630Crossref, Medline, Google Scholar
52 Nalbantoglu J, Gilfix BM, Bertrand P, et al: Predictive value of apolipoprotein E genotyping in Alzheimer's disease: results of an autopsy series and an analysis of several combined studies. Ann Neurol 1994; 36:889–895Crossref, Medline, Google Scholar
53 Corder EH, Saunders AM, Risch NH, et al: Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer's disease. Nat Genet 1994; 7:180–184Crossref, Medline, Google Scholar
54 Hyman BT, Gomez-Isla T, Briggs M, et al: Apolipoprotein E and cognitive change in an elderly population. Ann Neurol 1996; 40:55–66Crossref, Medline, Google Scholar
55 Blacker D, Haines JL, Rodes L, et al: ApoE-4 and age at onset of Alzheimer's disease: the NIMH genetics initiative. Neurology 1997; 48:139–147Crossref, Medline, Google Scholar
56 Roses AD, Strittmatter WJ, Pericak-Vance MA, et al: Clinical application of apolipoprotein E genotyping to Alzheimer's disease. Lancet 1994; 343:1564–1565Crossref, Medline, Google Scholar
57 Schmechel DE, Saunders AM, Strittmatter WJ, et al: Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer's disease. Proc Natl Acad Sci USA 1993; 90:9649–9653Crossref, Medline, Google Scholar
58 Strittmatter WJ, Saunders AM, Schmechel D, et al: Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer's disease. Proc Natl Acad 1993; 90:1977–1981Crossref, Medline, Google Scholar
59 Strittmatter WJ, Weisgraber KH, Huang DY, et al: Binding of human apolipoprotein E to synthetic amyloid beta peptide: isoform-specific effects and implications for late-onset Alzheimer's disease. Proc Natl Acad 1993; 90:8098–8102Crossref, Medline, Google Scholar
60 Evans KC, Berger EP, Cho CG, et al: Apolipoprotein E is a kinetic but not a thermodynamic inhibitor of amyloid formation: implications for the pathogenesis and treatment of Alzheimer's disease. Proc Natl Acad Sci USA 1995; 92:763–767Crossref, Medline, Google Scholar
61 Ma J, Yee A, Brewer B Jr, et al: Amyloid-associated proteins alpha 1-antichymotrypsin and apolipoprotein E promote assembly of Alzheimer beta-protein into filaments. Nature 1994; 372:92–94Crossref, Medline, Google Scholar
62 Strittmatter WJ, Saunders AM, Goedert M, et al: Isoform-specific interactions of apolipoprotein E with microtubule-associated protein tau: implications for Alzheimer's disease. Proc Natl Acad Sci USA 1994; 91:11183–11186Crossref, Medline, Google Scholar
63 Roses AD, Einstein G, Gilbert J, et al: Morphological, biochemical, and genetic support for an apolipoprotein E effect on microtubular metabolism. Ann NY Acad Sci 1996; 777:146–157Crossref, Medline, Google Scholar
64 Nathan BP, Chang KC, Bellosta S, et al: The inhibitory effect of apolipoprotein E4 on neurite outgrowth is associated with microtubule depolymerisation. J Biol Chem 1995; 270:19791–19799Crossref, Medline, Google Scholar
65 Mahley RW, Nathan BP, Pitas RE: Apolipoprotein E: structure, function, and possible roles in Alzheimer's disease. Ann NY Acad Sci 1996; 777:139–145Crossref, Medline, Google Scholar
66 Yankner BA, Duffy LK, Kirschner DA: Neurotrophic and neurotoxic effects of amyloid beta protein: reversal by tachykinin neuropeptides. Science 1990; 250:279–282Crossref, Medline, Google Scholar
67 Lorenzo A, Yankner B: Beta-amyloid neurotoxicity requires fibril formation and is inhibited by Congo red. Proc Natl Acad Sci USA 1994; 91:12243–12247Crossref, Medline, Google Scholar
68 Loo DT, Copani A, Pike CJ, et al: Apoptosis is induced by beta-amyloid in cultured central nervous system neurons. Proc Natl Acad Sci USA 1993; 90:7951–7955Crossref, Medline, Google Scholar
69 Busciglio J, Lorenzo A, Yeh J, et al: Beta amyloid fibrils induce tau phosphorylation and loss of microtubule binding. Neuron 1995; 14:879–888Crossref, Medline, Google Scholar
70 Terry RT: The pathogenesis of Alzheimer's disease: an alternative to the amyloid hypothesis. J Neuropathol Exp Neurol 1996; 55:1023–1025Crossref, Medline, Google Scholar
71 Sohal RS, Weindruch R: Oxidative stress, caloric restriction, and aging. Science 1996; 273:59–63Crossref, Medline, Google Scholar
72 Smith CD, Carney JM, Starke-Reed PE, et al: Excess brain protein oxidation and enzyme dysfunction in normal aging and in Alzheimer's disease. Proc Natl Acad Sci USA 1991; 88:10540–10543Crossref, Medline, Google Scholar
73 Subbarao KV, Richardson JS, Ang LC: Autopsy samples of Alzheimer's cortex show increased peroxidation in vitro. J Neurochem 1990; 55:342–345Crossref, Medline, Google Scholar
74 Pappolla MA, Omar RA, Kim KS, et al: Immunohistochemical evidence of oxidative stress in Alzheimer's disease. Am J Pathol 1992; 140:621–628Medline, Google Scholar
75 Mark RJ, Hensley K, Butterfield DA, et al: Amyloid beta-peptide impairs ion-motive ATPase activities: evidence for a role in loss of neuronal Ca2+ homeostasis and cell death. J Neurosci 1995; 15:6239–6249Crossref, Medline, Google Scholar
76 Mattson MP, Rydel RE: Amyloid ox-tox transducers. Nature 1996; 382:674–675Crossref, Medline, Google Scholar
77 Disterhoft JF, Moyer JR Jr, Thompson LT: The calcium rationale in aging and Alzheimer's disease. Ann NY Acad Sci 1994; 747:382–406Crossref, Medline, Google Scholar
78 Fritze J, Walden J: Clinical findings with nimodipine in dementia: test of the calcium hypothesis. J Neural Transm 1995; 46(suppl):439–453Google Scholar
79 Yan SD, Chen X, Fu J, et al: RAGE and amyloid-beta peptide neurotoxicity in Alzheimer's disease. Nature 1996; 382:685–691Crossref, Medline, Google Scholar
80 Hori O, Brett J, Slattery T, et al: The receptor for advance glycation end products (RAGE) is a cellular binding site for amphoterin. J Biol Chem 1995; 270:25752–25761Crossref, Medline, Google Scholar
81 Sano M, Ernesto C, Thomas RG, et al: A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer's disease. N Engl J Med 1997; 336:1216–1222Crossref, Medline, Google Scholar
82 Rosen WG, Mohs RC, Davis KL: A new rating scale for Alzheimer's disease. Am J Psychiatry 1984; 141: 1356–1364Google Scholar
83 Schneider LS, Tariot PN, Goldstein B: Therapy with l-deprenyl (selegiline) and relation to abuse liability. Clin Pharmacol Ther 1994; 56:750–756Crossref, Medline, Google Scholar
84 Freedman M, Rewilak D, Xerri T, et al: l-deprenyl in Alzheimer's disease. Neurology 1998; 50:660–668Crossref, Medline, Google Scholar
85 Riekkinen P, Koivisto K, Helkala E, et al: Long-term double-blind trial of selegiline in Alzheimer's disease. Neurobiol Aging 1996; 15(suppl 1):S67Google Scholar
86 Burke WJ, Roccaforte WH, Wengel SP, et al: l-deprenyl in the treatment of mild dementia of the Alzheimer type: results of a 15-month trial. J Am Geriatr Soc 1993; 41:1219–1225Crossref, Medline, Google Scholar
87 Dysken MW, Mendels J, LeWitt P, et al: Milacemide: a placebo-controlled study in senile dementia of the Alzheimer type. J Am Geriatr Soc 1992; 40:503–506Crossref, Medline, Google Scholar
88 Orgogozo JM, Dartigues JF, Lafont S, et al: Wine consumption and dementia in the elderly: a prospective community study in the Bordeaux area. Rev Neurol (Paris) 1997; 153:185–192Medline, Google Scholar
89 Maxwell S, Cruickshank A, Thorpe G: Red wine and antioxidant activity in serum. Lancet 1994; 344:193–194Crossref, Medline, Google Scholar
90 Le Bars PL, Katz MM, Berman N, et al: A placebo-controlled, double-blind, randomized trial of an extract of ginkgo biloba for dementia. JAMA 1997; 278:1327–1332Crossref, Medline, Google Scholar
91 Pappolla MA, Sos M, Omar RA, et al: Melatonin prevents death of neuroblastoma cells exposed to the Alzheimer amyloid peptide. J Neurosci 1997; 17:1683–1690Crossref, Medline, Google Scholar
92 Mishima K, Okawa M, Hishikawa Y, et al: Morning bright light therapy for sleep and behavior disorders in elderly patients with dementia. Acta Psychiatr Scand 1994; 89:1–7Crossref, Medline, Google Scholar
93 Tohgi H, Abe T, Takahashi S: Concentration of serotonin and its related substances in the cerebrospinal fluid in patients with Alzheimer type dementia. Neurosci Lett 1992; 141:9–12Crossref, Medline, Google Scholar
94 McGeer PL, McGeer E, Rogers J, et al: Anti-inflammatory drugs and Alzheimer's disease. Lancet 1990; 335:1037Crossref, Medline, Google Scholar
95 McGeer PL, McGeer EG: Complement proteins and complement inhibitors in Alzheimer's disease. Res Immunol 1992; 143:621–624Crossref, Medline, Google Scholar
96 Rogers J, Schultz J, Brachova L, et al: Complement activation and beta-amyloid-mediated neurotoxicity in Alzheimer's disease. Res Immunol 1992; 143:624–630Crossref, Medline, Google Scholar
97 Aisen PS, Davis KL: Inflammatory mechanisms in Alzheimer's disease: implication for therapy. Am J Psychiatry 1994; 151:1105–1113Crossref, Medline, Google Scholar
98 Wyss-Coray T, Masliah E, Mallory M, et al: Amyloidogenic role of cytokine TGF-beta 1 in transgenic mice and in Alzheimer's disease. Nature 1997; 389:603–606Crossref, Medline, Google Scholar
99 Payami H, Schellenberg GD, Zareparsi S, et al: Evidence for association of HLA-A2 allele with onset age of Alzheimer's disease. Neurology 1997; 49:512–518Crossref, Medline, Google Scholar
100 Jenkinson ML, Bliss MR, Brain AT, et al: Rheumatoid arthritis and senile dementia of the Alzheimer's type. Br J Rheumatol 1989; 28:86–88Crossref, Medline, Google Scholar
101 Breitner JCS, Gau BA, Welsh KA, et al: Inverse association of anti-inflammatory treatments and Alzheimer's disease: initial results of a co-twin control study. Neurology 1994; 44:227–232Crossref, Medline, Google Scholar
102 Andersen K, Launer LJ, Ott A, et al: Do nonsteroidal anti-inflammatory drugs decrease the risk for Alzheimer's disease? The Rotterdam study. Neurology 1995; 45:1441–1445Crossref, Medline, Google Scholar
103 Stewart WF, Kawas C, Corrada M, et al: Risk of Alzheimer's disease and duration of NSAID use. Neurology 1997; 48:626–632Crossref, Medline, Google Scholar
104 Rich JB, Rasmusson DX, Folstein MF, et al: Nonsteroidal anti-inflammatory drugs in Alzheimer's disease. Neurology 1995; 45:51–55Crossref, Medline, Google Scholar
105 Rogers J, Kirby LC, Hempelman SR, et al: Clinical trial of indomethacin in Alzheimer's disease. Neurology 1993; 43:1609–1611Crossref, Medline, Google Scholar
106 Aisen PS, Davis KL: The search for disease-modifying treatment for Alzheimer's disease. Neurology 1997; 48(suppl 6):S35–S41Google Scholar
107 MacKenzie IRA: Antiinflammatory drugs in the treatment of Alzheimer's disease. J Rheumatol 1996; 23:806–808Medline, Google Scholar
108 El Khoury J, Hickman SE, Thomas CA, et al: Scavenger receptor-mediated adhesion of microglia to beta-amyloid fibrils. Nature 1996; 382:716–719Crossref, Medline, Google Scholar
109 Pericak-Vance MA, Bass MP, Yamaoka LH, et al: Complete genomic screen in late-onset familial Alzheimer's disease. JAMA 1997; 278:1237:1241Crossref, Medline, Google Scholar
110 Kamboh MI, Sanghera DK, Ferrel RE, et al: Apo-E4 associated Alzheimer's disease risk is modified by alpha-1-antichymotrypsin polymorphism. Nat Genet 1995; 10:486–488Crossref, Medline, Google Scholar
111 Kang DE, Saitoh T, Chen X, et al: Genetic association of the low-density lipoprotein gene (LRP), an apolipoprotein E receptor, with late-onset Alzheimer's disease. Neurology 1997; 49:56–61Crossref, Medline, Google Scholar
112 Simpkins JW, Singh M, Bishop J: The potential role for estrogen replacement therapy in the treatment of the cognitive decline and neurodegeneration associated with Alzheimer's disease. Neurobiol Aging 1994; 15(suppl 2):S195–S197Google Scholar
113 Henderson VW, Paganini-Hill A, Emanuel CK, et al: Estrogen replacement therapy in older women: comparisons between Alzheimer's disease cases and non-demented control subjects. Arch Neurol 1994; 51:896–900Crossref, Medline, Google Scholar
114 Mortel KF, Meyer JS: Lack of postmenopausal estrogen replacement therapy and the risk of dementia. J Neuropsychiatry Clin Neurosci 1995; 7:334–337Link, Google Scholar
115 Tang MX, Jacobs D, Stern Y, et al: Effect of oestrogen during menopause on risk and age at onset of Alzheimer's disease. Lancet 1996; 348:429–432Crossref, Medline, Google Scholar
116 Kawas C, Resnick S, Morrison A, et al: A prospective study of estrogen replacement therapy and the risk of developing Alzheimer's disease: the Baltimore Longitudinal Study of Aging. Neurology 1997; 48:1517–1521Crossref, Medline, Google Scholar
117 Schneider LS, Farlow MR, Henderson VW, et al: Effects of estrogen replacement therapy on response to tacrine in patients with Alzheimer's disease. Neurology 1996; 46:1580–1584Crossref, Medline, Google Scholar
118 Kesner RP: Reevaluation of the contribution of the basal forebrain cholinergic system to memory. Neurobiol Aging 1988; 9:609–616Crossref, Medline, Google Scholar
119 Cummings JL, Kaufer D: Neuropsychiatric aspects of Alzheimer's disease: the cholinergic hypothesis revisited. Neurology 1996; 47:876–883Crossref, Medline, Google Scholar
120 Bowen DM, Smith CB, White P, et al: Neurotransmitter-related enzymes and indices of hypoxia in senile dementia and other abiotrophies. Brain 1976; 99:459–496Crossref, Medline, Google Scholar
121 Dekosky ST, Scheff SW, Markesbery WR: Laminar organization of cholinergic circuits in human frontal cortex in Alzheimer's disease and aging. Neurology 1985; 35:1425–1431Crossref, Medline, Google Scholar
122 Davies P, Maloney AJF: Selective loss of central cholinergic neurons in Alzheimer's disease (letter). Lancet 1976; 2:1403Crossref, Medline, Google Scholar
123 Whitehouse PJ, Price DL, Struble RG, et al: Alzheimer's disease and senile dementia: loss of neurons in the basal forebrain. Science 1982; 215:1237–1239Crossref, Medline, Google Scholar
124 Perry EK, Tomlinson BE, Blessed G, et al: Correlations of cholinergic abnormalities with senile plaques and mental test scores in senile dementia. BMJ 1978; 2:1457–1459Crossref, Medline, Google Scholar
125 Francis PT, Palmer AM, Sims NR, et al: Neurochemical studies of early-onset Alzheimer's disease. Possible influence on treatment. N Engl J Med 1985; 313:7–11Crossref, Medline, Google Scholar
126 Sims NR, Bowen DM, Smith CCT, et al: Glucose metabolism and acetylcholine synthesis in relation to neuronal activity in Alzheimer's disease. Lancet 1980; 1:333–336Crossref, Medline, Google Scholar
127 Wallace WC, Lieberburg I, Schenk D, et al: Chronic elevation of secreted amyloid precursor protein in subcortically lesioned rats, and its exacerbation in aged rats. J Neurosci 1995; 15:4896–4905Crossref, Medline, Google Scholar
128 Stern RG, Davis KL: Research in treating cognitive impairment in Alzheimer's disease, in The Dementias: Diagnosis, Management, and Research, 2nd edition, edited by Weiner MF. Washington, DC, American Psychiatric Press, 1996, pp 331–353Google Scholar
129 Bodick NC, Offen WW, Levey AI, et al: Effects of xanomeline, a selective muscarinic receptor agonist, on cognitive function and behavioral symptoms of Alzheimer's disease. Arch Neurol 1997; 54:465–473Crossref, Medline, Google Scholar
130 Davis KL, Mohs RC: Enhancement of memory processes in Alzheimer's disease with multiple-dose intravenous physostigmine. Am J Psychiatry 1982; 139:1421–1424Crossref, Medline, Google Scholar
131 Mohs RC, Davis KL: The experimental pharmacology of Alzheimer's disease and related dementias, in Psychopharmacology: The Third Generation of Progress, edited by Meltzer HY, New York, Raven Press, 1987, pp 921–928Google Scholar
132 Summers WK, Majovski LV, Marsh GM, et al: Oral tetrahydroaminoacridine in long-term treatment of senile dementia, Alzheimer type. N Engl J Med 1986; 315:1241–1245Crossref, Medline, Google Scholar
133 Davis KL, Thal LJ, Gamzu ER, et al: A double-blind, placebo-controlled multicenter study of tacrine for Alzheimer's disease. N Engl J Med 1992; 327:1253–1259Crossref, Medline, Google Scholar
134 Farlow M, Gracon SI, Hershey LA, et al: A controlled trial of tacrine in Alzheimer's disease. JAMA 1992; 268:2523–2529Crossref, Medline, Google Scholar
135 Knapp MJ, Knopman DS, Solomon PR, et al: A 30-week randomized controlled trial of high-dose tacrine in patients with Alzheimer's disease. JAMA 1994; 271:985–991Crossref, Medline, Google Scholar
136 Knopman D, Schneider L, Davis K, et al: Long-term tacrine (Cognex) treatment: effects on nursing home placement and mortality. Neurology 1996; 47:166–177Crossref, Medline, Google Scholar
137 Watkins PB, Zimmerman HJ, Knapp MJ: Hepatotoxic effects of tacrine administration in patients with Alzheimer's disease. JAMA; 1994:992–998Google Scholar
138 Rogers SL, Friedhoff LT, and the Donepezil Study Group: The efficacy and safety of donepezil in patients with Alzheimer's disease: results of a US multicenter, randomized, double-blind, placebo-controlled trial. Dementia 1996; 7:293–303Medline, Google Scholar
139 Rogers SL, Farlow MR, Doody RS, et al: A 24-week, double-blind, placebo-controlled trial of donepezil in patients with Alzheimer's disease. Neurology 1998; 50:136–145Crossref, Medline, Google Scholar
140 Bondareff W, Mountjoy CQ, Roth M: Loss of neurons of origin of the adrenergic projection to the cerebral cortex (nucleus locus ceruleus) in senile dementia. Neurology 1982; 32:164–168Crossref, Medline, Google Scholar
141 Zweig RM, Ross CA, Hedreen JC, et al: The neuropathology of aminergic nuclei in Alzheimer's disease. Ann Neurol 1988; 24:233–242Crossref, Medline, Google Scholar
142 Zubenko GS, Moossy J: Major depression in primary dementia: clinical and neuropathologic correlates. Arch Neurol 1988; 45:1182–1186Crossref, Medline, Google Scholar

