
Reduced Hippocampal Volume in Association With P50 Nonsuppression Following Traumatic Brain Injury
Abstract
Traumatic brain injury (TBI) may produce persistently impaired auditory gating. This cholinergic-dependent, hippocampally mediated preattentive cognitive function that facilitates filtering of auditory stimuli may be indexed by the P50 evoked waveform to paired auditory stimuli. Abnormal P50 suppression post TBI is believed to result from injury to the hippocampus and/or its afferent cholinergic projections. This hypothesis was tested by comparing hippocampal and total brain volumes on MRI between ten P50-nonsuppressing TBI patients and ten normal control subjects matched for age, gender, and education. TBI subjects had highly significant bilateral hippocampal volume reductions, even when covaried for reductions in total brain volume. Degree of volume loss was not correlated with initial TBI severity. Findings support the hypothesis that hippocampal injury underlies P50 nonsuppression post TBI and suggest that such structural abnormalities may be observed even in “mildly” injured persons.
Traumatic brain injury (TBI), at all levels of initial severity, may produce persistent neuropsychiatric symptoms,1–3 including impairment of auditory gating.4,5 Auditory gating is a pre-attentive cognitive function that facilitates selective attention by providing a mechanism for filtering out (gating) irrelevant or excessive auditory stimuli.6 It is largely dependent on the ability of cholinergic afferents from basal forebrain cholinergic nuclei (Ch1 and Ch2)6–10 to effect multimodal sensory (including auditory) gating within the hippocampus. Cholinergic projections to the hippocampus are particularly vulnerable to disruption by TBI.4,11–13 Additionally, hippocampal cortex may be particularly vulnerable to direct biomechanical injury during TBI.14,15 Together, these factors may combine to produce impairment of hippocampal functioning after TBI, the consequences of which include impairments in auditory gating.
Auditory gating may be indexed by the P50 evoked response to paired auditory stimuli.6,7,16 The amplitude of the P50 evoked responses to these paired auditory stimuli (which consist of a conditioning and a test click) are used to construct a ratio (P50 ratio) of the ability to inhibit, or gate, auditory cortical responses.8,10 When the P50 ratio is abnormally high (P50 nonsuppression), it suggests that the hippocampus is unable to adequately inhibit responses to repetitive auditory stimuli. Clinically, this form of impaired inhibition is manifest as impaired auditory gating, a condition in which patients are unable to filter out irrelevant auditory stimuli and consequently fail to consistently mount robust selective attention.
We recently investigated the relationship between TBI, impaired auditory gating, and P50 suppression and demonstrated persistent P50 nonsuppression among TBI patients with symptoms of impaired auditory gating.5 We interpreted this finding as evidence of hippocampal cholinergic dysfunction following TBI, consistent with our previous speculation4 and echoing similar suggestions by other authors.11–13,17,18 These suggestions have been referred to as the cholinergic hypothesis of attention and memory impairment following TBI.4 This hypothesis suggests that cholinergic dysfunction may be produced by even relatively mild degrees of concussive injury and that persistent symptoms of impaired auditory gating following TBI may be understood, at least in part, as a consequence of hippocampal cholinergic dysfunction.
This hypothesis would be supported further by neuroimaging evidence of hippocampal pathology in TBI patients with symptoms of impaired auditory gating. Volumetric magnetic resonance imaging (MRI) may offer one such source of evidence. For example, Bigler et al.19 have demonstrated an association between verbal memory impairment and reduced hippocampal volume among patients with persistently impaired cognition following TBI. Although these authors do not frame their conclusions in the context of the cholinergic hypothesis, their results do suggest that volumetric MRI investigation of the hippocampus in TBI patients with persistent memory impairment may reveal important relationships between clinical presentation and significant structural changes in the brain. Their results further suggest that such methods may reveal abnormalities that might escape detection using more conventional methods of clinical neuroimaging.
More immediately relevant to the cholinergic hypothesis, Waldo et al.20 demonstrated an association between P50 nonsuppression and reduced hippocampal volume on MRI among patients with schizophrenia. P50 nonsuppression in schizophrenia is believed to reflect hippocampal cholinergic dysfunction due to a genetically determined postsynaptic defect in the alpha-7 nicotinic receptor of hippocampal GABA interneurons6,7—a fundamentally different mechanism from the cholinergic deafferentation and/or cortical injury that we suggest is the basis for this finding following TBI. Nonetheless, their observation has been used to offer support for the suggestion that P50 nonsuppression is predicated on hippocampal abnormalities.
Given these observations, we speculated that volumetric MRI analysis of the hippocampus in patients with P50 nonsuppression and impaired auditory gating following TBI might be similarly revealing. We felt that if reduced volume was observed it might lend even stronger support for the hippocampal basis of P50 nonsuppression in humans than had been provided in the Waldo et al. study alone.20 Additionally, we believed that demonstration of a structural abnormality in the area that we hypothesize is the primary generator of the P50 evoked response would provide more specific support for the hypothesis that impaired auditory gating following TBI is predicated on hippocampal cholinergic dysfunction. We therefore undertook the present study to investigate the hypothesis that impaired auditory gating and P50 nonsuppression following TBI are associated with reduced hippocampal volume on magnetic resonance imaging.
METHODS
Subjects
Subjects were recruited from the first author's neuropsychiatry clinic, from colleagues in the community, and through local newspaper advertisements. The recruitment targeted persons who had experienced a TBI at least one year prior to participation and who had developed persistently impaired attention and memory. An initial telephone screening interview excluded persons with a history of pre-TBI psychiatric, neurologic, or substance (including alcohol) problems, any history of seizures, and current use of medications known to affect P50 suppression.21
Qualifying subjects gave informed consent in accordance with the guidelines of our institutional review board. Information about TBI was retrospectively assessed on the basis of patient/family interview and/or record review. Subjects with nonpenetrating TBI producing at least 15 minutes of posttraumatic amnesia (PTA), not requiring neurosurgical intervention, occurring at least one year prior to interview, and resulting in symptoms consistent with impaired auditory gating were invited to participate in the study. This method of determining TBI severity was chosen because estimation of duration of PTA was more consistently available than Glasgow Coma Scale22 scores, particularly for patients with milder initial injuries.
Subjects were interviewed on the basis of DSM-IV23 criteria to better characterize pre- and post-TBI psychiatric history, and the results of this interview were used to exclude patients with any identifiable Axis I disorder (including substance abuse/dependence) prior to their TBI, significant post-TBI substance abuse/dependence (including alcohol), and any active Axis I mood, anxiety, or substance disorder at the time of P50 recording and MRI.
All subjects had Glasgow Outcome Scale (GOS)24 scores of 5 (“good” recovery) and age- and education-adjusted Mini-Mental State Examination (MMSE) scores at or above the 25th percentile (in general, this meant MMSE score ≥28/30). Although neither GOS nor MMSE offers a particularly sensitive method of excluding the possibility of persistent problems due to TBI, our purpose in using these measures as inclusion/exclusion criteria was to define a population that, regardless of initial injury severity, experiences the “good” outcome and the more subtle types of neurocognitive impairment that sometimes results in description of these patients as the “walking wounded” or as those with “invisible injury.” Additionally, these entrance requirements were established to better ensure TBI subjects' ability to competently consent to participate in the study. There was one exception to the minimum MMSE score requirement: a 46 year-old male subject with a remote history of severe TBI (10 years earlier), who scored at the 10th percentile on the MMSE (transposed two letters in “world” backwards, missed date, and missed two items on spontaneous recall; however, cued recall was normal) but who otherwise fit the intended clinical picture of the study group, was included.
Ten control subjects on whom MRI of the brain had been obtained as part of an earlier study were selected for comparison with the study group. These comparison subjects had no history of traumatic brain injury, met Research Diagnostic Criteria for Never Mentally Ill, and were matched by gender, education, and within one year of age to the TBI subjects.
P50 Recording
P50 recordings were obtained on these TBI subjects according to the methods and criteria we have previously described.4,5,8,10 Evoked potential recordings were performed with the subject alert, supine, and with eyes fixed on a distant target. Evoked responses were recorded by using a gold disk electrode at the vertex, referenced to one ear, and eye movements were simultaneously recorded using an electrode placed between the right superior orbit and the lateral canthus. Auditory stimuli (1 ms duration, 20–12 kHz) were presented via headphones at 30–45 dB above hearing threshold in pairs (0.5-s intrapair interval, 10-s interstimulus interval) in a conditioning-testing design. Three sets of average responses were constructed, each based on responses to 16 pairs of stimuli, and used to create an overall grand average of the conditioning and test P50 responses for each subject.
The P50 wave was digitally filtered, identified, and measured in each set of averages, using a previously described computer algorithm.8,25 The filtering algorithm identified the conditioning P50 wave as the most positive peak between 40 and 80 ms after the first stimulus. P50 amplitude was measured relative to the preceding negativity, and a conditioning wave amplitude of at least 0.5 μV was required for the trial to be considered valid. The test P50 wave was identified as the most positive peak with a latency from the test stimulus within 10 ms of the latency of the conditioning P50 response. The amplitude of the test P50 wave divided by the amplitude of the conditioning P50 wave, expressed as a percentage and referred to here as the “P50 ratio,” was used as a measure of auditory gating. P50 nonsuppression was defined as a P50 ratio of greater than 60. TBI subjects were required to demonstrate P50 nonsuppression in order to qualify for volumetric MRI assessment.
Volumetric MRI Assessment
MRI of the brain was performed at the Denver Veterans Affairs Medical Center on a Phillips Gyroscan 1.5-T system. A standard head coil using a 3-D spoiled gradient echo acquisition was used to obtain a series of 124 1.7-mm thick, T1-weighted coronal images of the head, with TR/TE of 40/5 ms and a 40° flip-angle. A 256×256 matrix acquisition produced voxel dimensions of 0.94×0.94×1.7 mm.
Image Reconstruction and Region of Interest Definition
Image reconstruction and segmentation was performed by using a locally modified, semiautomated, manual segmentation routine designed in Interactive Data Language 4.0.1 (Research Systems, Boulder, CO). The volume computation algorithm based on these images has been found to be accurate to within ±1% through testing on regular and irregular phantoms of known volume.
All MRI files were stripped of all identifying information to permit segmentation blind to subject condition. Using previously described methods,26 the total brain volume was determined via manual segmentation into CSF and gray matter from the coronal images. A single investigator (J.T.) performed all whole-brain volume determinations. Intrarater reliability on these measurements, determined by repeat segmentation of five randomly selected brains, was 0.94 (Pearson product-moment correlation, P<0.05).
The MRI area delimiting the hippocampus was established by using landmarks determined by consensus of the authors (D.A., M.R., D.R., E.S.), the use of neuroanatomical atlases,27,28 and the results of previously described methods.19,29,30 The retrocommissural hippocampus,28 its most morphologically developed and easily visualized portion on MRI, was the ROI defined for volumetric analysis (Figure 1). Right and left hippocampal volumes were determined by manual segmentation performed by a single investigator (D.A.) blind to subject condition. Intrarater reliability using this method, determined by repeated segmentation of five randomly selected hippocampi, was 0.99 (Pearson product-moment correlation, P<0.05).
Qualitative validity of these methods was determined by inspection of three-dimensional reconstructions of the segmented volumes using Tecplot 7.0 (Amtec Engineering, Inc., Bellevue, WA), to verify that the method described provided a valid method of segmenting the hippocampus (Figure 2).
Statistical Analyses
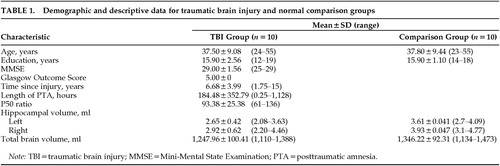
Statistica 5.1 (StatSoft, Tulsa, OK) was used for all analyses. Descriptive statistics regarding age, education, MMSE, GOS, time elapsed since TBI, length of posttraumatic amnesia (PTA), P50 ratio, total brain volume, and right and left hippocampal volume for the TBI and comparison groups are included in Table 1. Pearson product-moment correlations were performed on the data of all subjects to look for significant correlations between the MRI volumetric variables (total brain, right, and left hippocampal volumes) and age, gender, and education to determine whether any of the demographic variables warranted inclusion as covariates in the volumetric analyses. No significant correlations were found.
A one-way analysis of variance (ANOVA), with group (TBI vs. comparison) as the independent variable and total brain volume as the dependent variable, was performed to determine the significance of the effect of group on total brain volume. The effect of group on hippocampal volumes was then evaluated by using a two-way analysis of covariance (ANCOVA), with group (TBI vs. comparison) as the independent variable, hippocampal volume as the dependent variable, and total brain volume as a fixed covariate (based on results of the ANOVA for group by total brain volume); hemisphere was included as a repeated measure. Planned comparisons were performed to test the hypotheses that 1) right hippocampal volume is reduced in the TBI group compared with the normal group; and 2) left hippocampal volume is reduced in the TBI group compared with the normal group.
Preliminary analyses of the effects of initial injury severity on total brain and hippocampal volumes were also performed. Our previous findings demonstrated no significant effect of injury severity (mild/moderate vs. severe) on P50 suppression, with both mild/moderate and severe TBI subjects demonstrating significant P50 ratio differences compared with normal subjects but not compared with each other.5 For this preliminary analysis, we hypothesized that we would observe a similar effect of injury severity on hippocampal volume. Given the relatively small sample size of the present study, we concluded that it would be difficult to perform a mean ingful analysis of the effect of injury severity on total brain or hippocampal volume by transforming duration of PTA into categorical variables (mild, moderate, severe) as was done in our previous study. Instead, we used duration of PTA as a continuous variable to perform a Pearson product-moment correlation between this variable and each of the volumetric variables under study.
RESULTS
Two-way ANCOVA demonstrated a highly significant effect of group on hippocampal volume (F=11.9, df=1,17, P<0.003; Figure 3), with reduction of hippocampal volume in the TBI group. Planned comparisons demonstrated that both right and left hippocampal volumes in the TBI group are highly significantly less than those of the normal comparison group (F=8.6, df=1,17, P<0.009, and F=15.6, df=1,17, P<0.001, respectively). Although there was an effect of group on total brain volume (smaller total brain volume in the TBI group, F=5.2, df=1,18, P<0.04), this effect did not diminish the significance of the effect of group on hippocampal volume. There was a significant hemisphere effect on hippocampal volume in both groups (F=28.4, df=1,16, P<0.001), left hippocampus being smaller than the right, but there was no interaction between group (TBI vs. comparison) and hemisphere, suggesting that the volume loss in the TBI group is relatively symmetric.
Duration of posttraumatic amnesia, taken as an indication of TBI severity, was not significantly correlated with total brain volume (r2=0.19, P=0.20), left hippocampal volume (r2=0.00, P=0.99), or right hippocampal volume (r2=0.00, P=0.89).
DISCUSSION
The present study was performed to investigate whether our hypothesis of hippocampal cholinergic dysfunction following TBI could be further supported with evidence of structural hippocampal abnormalities in TBI patients with symptoms of impaired auditory gating and P50 nonsuppression. The results of this study suggest a disproportionate effect of TBI on the hippocampus, with volume reduction in this structure being greater than can be accounted for by reduction in total brain volume. Disproportionate hippocampal volume loss among P50-nonsuppressing TBI patients provides support for the cholinergic hypothesis of auditory gating impairment following TBI by demonstrating convergent structural and electrophysiologic evidence of hippocampal abnormalities in these patients. Although the sample size in this study is relatively small, the effect size (ω2=0.31) derived from the data acquired suggests that this sample is large enough to permit a meaningful investigation of differences in hippocampal volume between these two groups, with power between 0.8 and 0.9.31 Additionally, the significance value of effect of group on hippocampal volume (P<0.003) suggests that the likelihood of a Type I statistical error is very low. Taken together, these analyses offer some reassurance that the findings may be regarded as robust even in the context of a relatively small study. To make the potential implications of these findings as clear as possible, we provide below a brief review of the cholinergic hypothesis.
The cholinergic hypothesis of attention and memory impairment due to TBI is based on the premise that selective and sustained attention derive from the function of and relationships among a selective distributed network of central nervous system structures.32 The network includes the reticular formation,33–35 thalamus,36,37 hippocampus and entorhinal areas,32,38 bifrontal and right parietal lobes,32,39 and axonal connections between these structures.32,40 Within this network, the hippocampus is required for sensory gating, the pre-attentive cortical process that facilitates the filtering of relevant highly processed multimodal sensory information from irrelevant background stimuli and the presentation of relevant information to the network for attentional and additional processing.8,32
The flow of highly processed sensory information through the hippocampus begins with its entry into the dentate gyrus and CA3 region, from whence it flows to the CA1 region and then to other subcortical and neocortical sites16,32,41,42 (Figure 4). As information enters the CA3 region it is directed not only through the direct afferent pathway to glutamatergic neurons, but also through a recurrent pathway that terminates on CA3–CA4 GABA interneurons. Activation of these GABA interneurons results in transmission of inhibitory signals to the CA3 glutamatergic neurons, momentarily preventing additional flow of sensory information through the direct pathway. Acetylcholine, via its action at low-affinity alpha-7 nicotinic receptors,43 provides an excitatory postsynaptic potential (EPSP) to these CA3–CA4 GABA interneurons, permitting their activation upon receipt of information from the recurrent pathway.6,7,16,16,44 Without cholinergic input to provide this EPSP, the GABA interneurons are not fully responsive to stimulation by incoming information from the recurrent pathway and therefore cannot provide adequate inhibition of subsequent incoming sensory input in the direct pathway. The cholinergically dependent momentary interruption of the flow of information through the hippocampus provided by these CA3-CA4 GABA interneurons is a principal means by which sensory filtering, or sensory gating, occurs.
Cholinergic afferents from the basal forebrain cholinergic nuclei (Ch1 and Ch2) project to the CA3-CA4 GABA interneurons in the hippocampus and appear to be particularly vulnerable to injury from even mild to moderate concussive forces.11–13,18,45–48 TBI may disrupt these cholinergic afferents through direct mechanical trauma,14,49–51 diffuse axonal injury,14,52–55 and/or disturbance of cholinergic function within the hippocampus.11,38,45,47 When cholinergic function in the hippocampus is disrupted because of TBI, impairments of auditory sensory gating and attention may develop and may be electrophysiologically indexed by nonsuppression of the P50 evoked response to paired auditory stimuli.4,5
Given this background information, the cholinergic hypothesis of attention and memory impairment following TBI states that 1) selective and sustained attention by the distributed attentional network are prerequisites for normal declarative/episodic memory; 2) pre-attentive filtering of highly processed sensory (including auditory) stimuli by the hippocampus is a prerequisite for the development of normal selective attention; 3) the process of auditory gating in the hippocampus is cholinergically dependent; 4) TBI disrupts hippocampal cholinergic function; and 5) therefore, TBI may impair attention and memory via disruption of the cholinergically dependent process of auditory gating.4,5
The patients studied here have symptoms of impaired auditory gating and P50 nonsuppression, both of which are consistent with the hypothesis of impaired hippocampal cholinergic function following TBI. The disproportionate hippocampal volume loss observed in these patients further supports this hypothesis by offering neuroimaging evidence of a structural hippocampal abnormality that converges with the symptoms and abnormal P50 suppression of these TBI patients. The hippocampal atrophy observed in these TBI patients most likely reflects a combination of deafferentation due to diffuse axonal injury and some degree of both direct and secondary cortical injury. These are predictable consequences of the biomechanical forces experienced during TBI.14,18,46,56,57 We suspect that diffuse axonal injury may contribute more strongly to the present finding. Reduced hippocampal volume measurable on MRI may reflect reduced white matter volume due to axonal losses. Although there may be secondary reduction in the number of hippocampal neurons following Wallerian degeneration of injured axons back to their somae in the hippocampus, the neurocircuitry described in Figure 4 suggests that injury to cholinergic fibers would more likely result in cell losses in the basal forebrain cholinergic nuclei than in the hippocampus itself. This interpretation is best supported by the ability of these patients to generate a P50 evoked potential (which requires hippocampal cortex) of reasonable amplitude despite hippocampal volume loss.
However, this study cannot clarify the relative contributions of cortical loss and white matter loss to the hippocampal volume loss observed here, so this interpretation must remain speculative at present. Additionally, and perhaps more troubling, this study cannot definitively determine whether the small hippocampal volume of these TBI patients is an effect of injury or is instead a premorbid risk factor that contributes to the development of auditory gating, attention, and memory impairments following TBI. Although the cholinergic hypothesis discussed herein suggests that the former possibility (hippocampal volume loss as an effect of injury) is more likely, additional studies are needed to further clarify this issue. These studies should employ methods such as proton magnetic resonance spectroscopy (e.g., using the NAA:Cr ratio to assess whether the hippocampus is small but composed of viable neurons or whether there is spectroscopic evidence of neuronal injury) and/or prospective (pre- and post-TBI) measurement of hippocampal volume. Further, information on the relationship between the present findings and attention and memory cannot be provided here because neuropsychological examination results relevant to this question are lacking. Further studies are needed to clarify the existence, nature, and strength of any such relationships.
Another interesting, although preliminary, result of this study is the finding of a relatively similar degree of hippocampal volume loss among all the TBI patients despite disparate initial TBI severity. It is important to restate that the TBI subjects in this study were included only if they had experienced relatively good global clinical outcome, had not developed cognitive impairments severe enough to warrant a dementia diagnosis, and generally appeared to be the sort of patients often described as the “walking wounded,” regardless of initial TBI severity. The intent of this sample design was to enable us to evaluate patients with respect to their long-term clinical outcome from TBI and to begin to address whether there are differences in hippocampal volume between persistently symptomatic, P50-nonsuppressing TBI patients with initially mild/moderate versus those with severe injuries. Using this same sample design in our previous study of P50 suppression, we found no significant differences between 11 mild/moderate initial TBI and 9 severe initial TBI subjects, although the P50 ratios of all TBI subjects were highly significantly different from those of the normal comparison subjects.5 In other words, it appeared that merely having persistent symptoms of impaired auditory gating was a more robust indicator of the presence of this electrophysiologic abnormality than one would anticipate based on the initial injury severity alone.
The present results also appear to provide preliminary support for the idea that even in apparently “mildly” affected TBI patients (as judged by GOS and MMSE), the presence of symptoms that are referable to a specific underlying neurophysiology is a stronger indicator of the presence of structural brain abnormalities than would be expected based on the initial severity of TBI alone. However, a negative finding in a sample of this size does merit concern that we may lack adequate power to detect correlations, if they exist, between initial TBI severity (as assessed by duration of PTA) and the volumetric variables. These findings nonetheless do suggest that additional structural and/or functional neuroimaging and electrophysiologic studies of these “good outcome” but persistently symptomatic TBI patients are important to pursue further.

FIGURE 1. Regions of interest (ROI) used for segmentation of the hippocampus viewed coronally (left) and sagittally (right)Asymmetry in the ROI outlined in the coronal image is the result of slight rotation of the head in the image. The ROI boundaries include only the retrocommissural hippocampus, its most morphologically developed and easily visualized portion on MRI. The precommissural (in the caudal portion of the area subcallosa, rostral to the septum verum) and supracommissural (indusium griseum, extending dorsorostrally along the length of the corpus callosum) portions of the hippocampus were not included in this assessment.

FIGURE 2. Three-dimensional reconstruction of the hippocampus based on manual segmentation in the coronal plane(A) views the right and left hippocampi from an anterior perspective, and (B) views the hippocampi from the left sagittal perspective.

FIGURE 3. Right and left hippocampal volumes, TBI vsnormal comparison groups (two-way analysis of covariance with total brain volume as the covariate; F=11.9, df=1,17, P < 0.003). See text for detailed results. Mean (symbol), standard error (box), and standard deviation (whiskers) are shown.

FIGURE 4. A simplified illustration of the relationship of the hippocampal excitatory and inhibitory circuitry involved in auditory sensory gatingCholinergic afferents provide the CA3–CA4 GABA interneurons with excitatory postsynaptic potentials that permit their activation when information enters the indirect pathway. Without these cholinergic neurons, the indirect pathway does not function and gating is impaired. Adapted from Freedman et al.16
 |
1 Neppe VM, Goodwin GT: The neuropsychiatric evaluation of the closed head injury of transient type (CHIT), in Evaluation and Treatment of Mild Traumatic Brain Injury, edited by Varney N, Roberts R. Mahweh, NJ, Lawrence Erlbaum, 1999, pp 149-208Google Scholar
2 McAllister TW: Mild traumatic brain injury and the postconcussive syndrome, in Neuropsychiatry of Traumatic Brain Injury, edited by Silver JM, Yudofsky SC, Hales RE. Washington DC, American Psychiatric Press, 1994, pp 357-392Google Scholar
3 McAllister TW: Neuropsychiatric sequelae of head injuries. Psychiatr Clin North Am 1992; 15:395-413Crossref, Medline, Google Scholar
4 Arciniegas D, Adler L, Topkoff J, et al: Attention and memory dysfunction after traumatic brain injury: cholinergic mechanisms, sensory gating, and a hypothesis for further investigation. Brain Inj 1999; 13:1-13Crossref, Medline, Google Scholar
5 Arciniegas D, Olincy A, Topkoff J, et al: Impaired auditory gating and P50 nonsuppression following traumatic brain injury. J Neuropsychiatry Clin Neurosci 2000; 12:77-85Link, Google Scholar
6 Adler LE, Freedman R, Ross RG, et al: Elementary phenotypes in the neurobiological and genetic study of schizophrenia. Biol Psychiatry 1999; 46:8-18Crossref, Medline, Google Scholar
7 Adler LE, Olincy A, Waldo M, et al: Schizophrenia, sensory gating, and nicotinic receptors. Schizophr Bull 1998; 24:189-202Crossref, Medline, Google Scholar
8 Adler LE, Hoffer LD, Wiser A, et al: Normalization of auditory physiology by cigarette smoking in schizophrenic patients. Am J Psychiatry 1993; 150:1856-1861Google Scholar
9 Adler LE, Nagamoto H, Drebing C, et al: Impaired auditory sensory gating in patients with 20 years of combat post-traumatic stress disorder and depression. New Research Program and Abstracts, American Psychiatric Association 144th Annual Meeting, New Orleans, LA, May 1, 1991Google Scholar
10 Nagamoto HT, Adler LE, Waldo MC, et al: Sensory gating in schizophrenics and normal controls: effects of changing stimulation intervals. Biol Psychiatry 1989; 25:549-561Crossref, Medline, Google Scholar
11 Dixon CE, Hamm RJ, Taft WC, et al: Increased anticholinergic sensitivity following closed skull impact and controlled cortical impact traumatic brain injury in the rat. J Neurotrauma 1994; 11:275-287Crossref, Medline, Google Scholar
12 Dixon CE, Ma X, Marion DW: Reduced evoked release of acetylcholine in the rodent neocortex following traumatic brain injury. Brain Res 1997; 749: 127-130Google Scholar
13 Murdoch I, Perry EK, Court JA, et al: Cortical cholinergic dysfunction after human head injury. J Neurotrauma 1998; 15:295-305Crossref, Medline, Google Scholar
14 Cassidy JW: Neuropathology, in Neuropsychiatry of Traumatic Brain Injury, edited by Silver JM, Yudofsky SC, Hales RE. Washington, DC, American Psychiatric Press, 1994, pp 43-79Google Scholar
15 Thatcher RW, Camacho M, Salazar A, et al: Quantitative MRI of the gray-white matter distribution in traumatic brain injury. J Neurotrauma 1997; 14:1-14Crossref, Medline, Google Scholar
16 Freedman R, Adler LE, Bickford P, et al: Schizophrenia and nicotinic receptors. Harv Rev Psychiatry 1994; 2:179-192Crossref, Medline, Google Scholar
17 Schmidt RH, Grady MS: Loss of forebrain cholinergic neurons following fluid-percussion injury: implications for cognitive impairment in closed head injury. J Neurosurg 1995; 83:496-502Crossref, Medline, Google Scholar
18 Dixon CE, Bao J, Bergmann JS, et al: Traumatic brain injury reduces hippocampal high-affinity [3H]choline uptake but not extracellular choline levels in rats. Neurosci Lett 1994; 180:127-130Crossref, Medline, Google Scholar
19 Bigler ED, Blatter DD, Anderson CV, et al: Hippocampal volume in normal aging and traumatic brain injury. Am J Neuroradiol 1997; 18:11-23Medline, Google Scholar
20 Waldo MC, Cawthra E, Adler LE, et al: Auditory sensory gating, hippocampal volume, and catecholamine metabolism in schizophrenics and their siblings. Schizophr Res 1994; 12:93-106Crossref, Medline, Google Scholar
21 Nagamoto HT, Adler LE, Hea RA, et al: Gating of auditory P50 in schizophrenics: unique effects of clozapine. Biol Psychiatry 1996; 40:181-188Crossref, Medline, Google Scholar
22 Teasdale G, Jennett B: Assessment of coma and impaired consciousness: a practical scale. Lancet 1974; ii:81-84Google Scholar
23 American Psychiatric Association: Diagnostic and Statistical Manual of Mental Disorders, 4th edition. Washington DC, American Psychiatric Association, 1994Google Scholar
24 Jennett B, Bond M: Assessment of outcome after severe brain damage: a practical scale. Lancet 1975; i:480-484Google Scholar
25 Nagamoto HT, Adler LE, Waldo MC, et al: Sensory gating in schizophrenics and normal controls: effects of changing stimulation intervals. Biol Psychiatry 1989; 25:549-561Crossref, Medline, Google Scholar
26 Reite M, Adams M, Simon J, et al: Auditory M100 component 1: relationship to Heschl's gyri. Cogn Brain Res 1994; 2:13-20Crossref, Medline, Google Scholar
27 Jackson GD, Duncan JS (ed): MRI Neuroanatomy: A New Angle on the Brain. New York, Churchill Livingstone, 1996Google Scholar
28 Nieuwenhuys R, Voogd J, van Huijzen C (ed): The Human Central Nervous System: A Synopsis and Atlas. Berlin, Springer-Verlag, 1988Google Scholar
29 Watson C, Andermann F, Gloor P, et al: Anatomic basis of amygdaloid and hippocampal volume measurement by magnetic resonance imaging. Neurology 1992; 42:1743-1750Google Scholar
30 Mu Q, Xie J, Wen Z, et al: A quantitative MR study of the hippocampal formation, the amygdala, and the temporal horn of the lateral ventricle in healthy subjects 40 to 90 years of age. Am J Neuroradiol 1999; 20:207-211Medline, Google Scholar
31 Cohen J (ed): Statistical Power Analysis for the Behavioral Sciences. Hillsdale, NJ, Lawrence Erlbaum, 1988Google Scholar
32 Mesulam M-M: From sensation to cognition. Brain 1998; 121:1013-1052Google Scholar
33 Jellinger K, Seitelberger F: Protracted post-traumatic encephalopathy: pathology, pathogenesis and clinical implications. J Neurol Sci 1970; 10:51-94Crossref, Medline, Google Scholar
34 Salazar AM, Grafman JH, Vance SC: Consciousness and amnesia after penetrating head injury: neurology and anatomy. Neurology 1986; 36:178-187Crossref, Medline, Google Scholar
35 Watson MR, Fenton GW, McClelland RJ, et al: The post-concussional state: neurophysiologic aspects. Br J Psychiatry 1995; 167:514-521Crossref, Medline, Google Scholar
36 Naatanen R (ed): Attention and Brain Function. Hillsdale, NJ, Lawrence Erlbaum, 1992Google Scholar
37 Naatanen R: The orienting response theory: an integration of informational and energetical aspects of brain function, in Adaptation to Stress and Task Demands: Energetical Aspects of Human Information Processing, edited by Hockey RGJ, Gaillard AWK, Coles M. Dordrecht, Netherlands, Martinus Nijhoff, 1986, pp 91-111Google Scholar
38 Gorman LK, Fu K, Hovda DA, et al: Effects of traumatic brain injury on the cholinergic system in the rat. J Neurotrauma 1996; 13:457-463Crossref, Medline, Google Scholar
39 Mesulam M-M: Attention, confusional states, and neglect, in Principles of Behavioral Neurology, edited by Mesulam M-M. Philadelphia, FA Davis, 1985, pp 125-168Google Scholar
40 Filley CM: The behavioral neurology of cerebral white matter. Neurology 1998; 50:1535-1540Google Scholar
41 Adler LE, Hoffer LJ, Griffith J, et al: Normalization by nicotine of deficient auditory sensory gating in the relatives of schizophrenics. Biol Psychiatry 1992; 32:607-616Crossref, Medline, Google Scholar
42 Adler LE, Hoffer L, Nagamoto HT, et al: Yohimbine impairs P50 auditory sensory gating in normal subjects. Neuropsychopharmacology 1994; 10:249-257Crossref, Medline, Google Scholar
43 Freedman R, Wetmore C, Stromberg I, et al: Alpha-bungarotoxin binding to hippocampal interneurons: immunocytochemical characterization and effects on growth factor expression. J Neurosci 1993; 13:1965-1975Google Scholar
44 Cave CB, Squire LR: Intact verbal and nonverbal short-term memory following damage to the human hippocampus. Hippocampus 1992; 2:151-163Crossref, Medline, Google Scholar
45 Dixon CE, Ma X, Marion DW: Effects of CDP-Choline treatment on neurobehavioral deficits after TBI and on hippocampal and neocortical acetylcholine release. J Neurotrauma 1997; 14:161-169Crossref, Medline, Google Scholar
46 Ciallella JR, Yan HQ, Ma X, et al: Chronic effects of traumatic brain injury on hippocampal vesicular acetylcholine transporter and M2 muscarinic receptor protein in rats. Exp Neurol 1998; 152:11-19Crossref, Medline, Google Scholar
47 Dixon CE, Bao J, Johnson KM, et al: Basal and scopolamine-evoked release of hippocampal acetylcholine following traumatic brain injury in rats. Neurosci Lett 1995; 198:111-114Crossref, Medline, Google Scholar
48 Saija A, Robinson SE, Lyeth BG, et al: The effects of scopolamine and traumatic brain injury on central cholinergic neurons. J Neurotrauma 1988; 5:161-170Crossref, Medline, Google Scholar
49 Eisenberg HM, Levin HS: CT and MRI in mild to moderate head injury, in Mild Head Injury, edited by Levin HS, Eisenberg HM, Benton AL. Oxford, UK, Oxford University Press, 1989, pp 133-141Google Scholar
50 Strub RL, Black FW (ed): Neurobehavioral Disorders: A Clinical Approach. Philadelphia, FA Davis, 1988Google Scholar
51 Silver JM, Yudofsky SC, Hales RE (eds): Neuropsychiatry of Traumatic Brain Injury. Washington, DC, American Psychiatric Press, 1994Google Scholar
52 Alexander MP: Traumatic brain injury, in Psychiatric Aspects of Neurologic Disease, 2nd edition, edited by Benson DF, Blumer E. New York, Grune and Stratton, 1982, pp 219-248Google Scholar
53 Jane JA, Steward O, Gennarelli TA: Axonal degeneration induced by experimental noninvasive minor head injury. J Neurosurg 1985; 62:96-100Crossref, Medline, Google Scholar
54 Filley CM (ed): Neurobehavioral Anatomy. Niwot, CO, University Press of Colorado, 1995Google Scholar
55 Markowitsch HJ, Calabrese P, Haupts M, et al: Searching for the anatomical basis of retrograde amnesia. J Clin Exp Neuropsychol 1993; 15:947-67Crossref, Medline, Google Scholar
56 Halliday AL: Pathophysiology, in Traumatic Brain Injury, edited by Marion DW. New York, Thieme Medical, 1999, pp 29-38Google Scholar
57 Thatcher RW, Camacho M, Salazar A, et al: Quantitative MRI of the gray-white matter distribution in traumatic brain injury. J Neurotrauma 1997; 14:1-14Crossref, Medline, Google Scholar

