
The Neuropsychiatric Profile of Addison’s Disease: Revisiting a Forgotten Phenomenon
METHOD
A patient presenting with psychotic symptoms was referred to our psychiatry service from the emergency department. We diagnosed her with underlying Addison’s disease. We followed the patient throughout her stay in the hospital for both the psychiatry and neurology services, and saw the patient in follow-up 2 months after discharge in our outpatient neurology clinic.
We accessed the literature on the neuropsychiatric symptoms associated with Addison’s disease using MEDLINE. The search included English-language publications from 1966 through April 2005. Keywords used included Addison’s disease, autoimmune polyendocrinopathies, glucocorticoids, hydrocortisone, encephalopathy, neuropsychiatric symptoms, psychotic disorders, mental disorders, depression, and cognition disorders. In addition, we reviewed citations in relevant papers.
CASE REPORT
A 30-year-old Caucasian married female nurse with no prior psychiatric history was referred to our psychiatry service by the emergency room for agitation, aggression, disinhibition, and delusions that her husband was pregnant, stealing money, and committing adultery. At times she appeared to respond to auditory and visual hallucinations.
History of Presenting Illness
Two weeks prior to her presentation ( Figure 1 ), the patient had become unwell with fatigue, severe weight loss, nausea, and vomiting. She was assessed at a community hospital and diagnosed with dehydration, food poisoning, and a sinus infection. She was treated with intravenous fluids, amoxicillin, and magnesium supplements. Her dose of thyroid medication, which had previously been prescribed for hypothyroidism, was increased. One week prior to her presentation, the patient had largely recovered and traveled to a warm climate to attend a friend’s wedding. Three days prior to presentation, while still on vacation, the patient became paranoid about her husband’s fidelity. She was reassured and consoled by her husband and behaved normally for the remaining 2 days of the trip. While traveling home, however, she became agitated and aggressive. On the plane she began yelling at fellow passengers, accusing them of being pregnant. She again accused her husband of committing adultery, selling their house, and stealing her money. She believed that the TV show she was watching on the plane was a reality show about her life. At one point, she attempted to climb out of the airplane window. When the plane landed, she was taken immediately to the hospital emergency room by her family and referred to psychiatry by the emergency room physician.
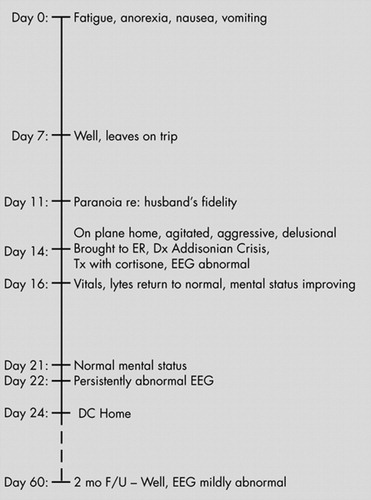
At presentation, the patient was febrile with a temperature of 97.9°F. She was hypotensive with a blood pressure of 76/48 and a pulse of 90 bpm. We were unable to obtain orthostatic blood pressure readings as she became very dizzy when standing. She was able to state here name, but was unaware of the date or that she was in a hospital. She was oriented to person but not to place or time. Her physical examination was unremarkable. She had a tan but also had increased pigmentation of her palmer creases, cheeks, upper lip, elbows, and knuckles. Although she had recently returned from a holiday in a sunny climate, her family felt that her pigmentation had been increasing over the previous year and was not simply a tan. Her family also reported she had lost 5 to 10 lbs in the preceding 2 weeks.
Past Medical History
The patient underwent surgery for Hirschsprung’s disease at 18 months of age. Apparently the surgery was complicated by an intraperitoneal infection which resulted in multiple adhesions, including adhesions to the ovaries and fallopian tubes. As a result, the patient had difficulty becoming pregnant and at age 26 underwent a tubal ligation and removal of the right ovary in preparation for in vitro fertilization, a procedure which had thus far been unsuccessful. She had required surgeries to drain cysts on her ovaries and underwent surgery for a bowel obstruction at age 29. She was diagnosed with hypothyroidism at age 28 and had been on thyroid supplementation since that time. As noted above, her dose of thyroid medication had recently been increased because of persisting fatigue.
The patient had no past psychiatric history. The only known history of psychiatric disturbance in her family was a paternal nephew who committed suicide. She was a nonsmoker who rarely consumed alcohol and did not use illegal substances or over-the-counter medications.
Investigations
The patient’s laboratory investigations are shown in Table 1 . Initial investigations revealed hyponatremia, negative plasma ethanol, and a negative urinalysis. A cranial computed tomography (CT) scan was normal. Her hyponatremia, hyperpigementation, hypotension, and history of thyroid disease led us to determine her serum cortisol level, which was undetectable. Subsequent investigations revealed further endocrine abnormalities, including low aldosterone and high ACTH, renin, growth hormone, prolactin, free T3, and thyroid peroxidase antibodies. The cortrosyn stimulation test was positive using a 250µg dose (pre-test cortisol<30nmol/liter, post-test (60-minute) cortisol<30nmol/liter). Levels of free T4, thyroid-stimulating hormone (TSH) and fasting glucose were normal.
 |
An EEG performed in the emergency room showed fluctuating levels of slow activity over both hemispheres without lateralizing features, lasting for 1 to 3 seconds. There was a mild and diffuse excess of slow activity over both hemispheres. A cranial magnetic resonance image (MRI) was normal.
Diagnosis
At the time of presentation, the patient’s history, physical examination, and laboratory findings suggested a diagnosis of an Addisonian crisis. Our main differential diagnoses included substance abuse, an infectious process or possibly a primary psychiatric disorder. Substance abuse was not supported by the patient’s history and was essentially ruled out with a negative urine toxicology screen. An infectious etiology was possible, particularly given the patient’s recent travel, so we requested an infectious disease consult. A primary psychiatric disorder was felt to be unlikely given the patient’s history, abnormal physical examination, laboratory findings, and largely negative personal and family history of psychiatric illness. She was also not noted to have any affective component to her illness and was not suicidal.
Course in Hospital
The patient was admitted with a diagnosis of Addisonian crisis and treated with intravenous normal saline and dexamethasone, supplemented by lorazepam for agitation. When the cortrosyn stimulation test was completed, dexamethasone was discontinued and replaced with a regimen of hydrocortisone. Fludrocortisone was added to her treatment 1 week later.
The patient’s vital signs, electrolytes, and free T4 level returned to baseline within 2 days. Her mental status improved steadily and returned to baseline within 1 week; however, she continued to notice subtle cognitive difficulties. A repeat EEG on her eighth day of admission was again abnormal, showing paroxysmal, poorly formed sharp and slow wave discharges over both central regions, more prominently on the left. After 10 days in the hospital, she was discharged and put on a regimen of hydrocortisone, fludrocortisone, and L -thyroxine.
Follow-Up
Two months after her admission, she reported feeling completely well physically and mentally. Her EEG had improved significantly but did show a single poorly formed sharp and slow wave discharge lasting for less than 1 second. Visual evoked potentials were within normal limits.
DISCUSSION
What Is the Nature of the Neuropsychiatric Presentation of Addison’s Disease?
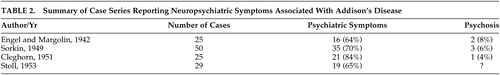
Four case series have documented mental status changes associated with cases of Addison’s disease ( Table 2 ). Engel and Margolin 3 in 1942 described 25 cases and documented a range of psychiatric symptoms in 64% of them. Mood and behavioral symptoms were most commonly reported, and psychosis was described in two patients. Sorkin, 4 in 1949, reported mental status changes in 70% of 50 Addison’s cases, and noted that mood and behavioral changes may be presenting features of the condition. He also reported three patients who developed psychosis. In 1951, Cleghorn 5 found apathy and negativism in 80% of 25 cases, with psychosis in one and seclusiveness, depression, and irritability occurring frequently. Stoll, in 1953, as reported by Smith et al., 6 found psychiatric symptoms in 65% of 29 cases. Mood symptoms and decreased motivation were common in less severe cases, and an “acute organic brain syndrome” was associated with severe cases. These series indicate, therefore, that mild disturbances in mood, motivation and behavior are core clinical features of Addison’s disease. Psychosis and extensive cognitive changes, including delirium, appear to occur more rarely, but are associated with severe disease and may be the presenting feature of Addisonian crisis.
 |
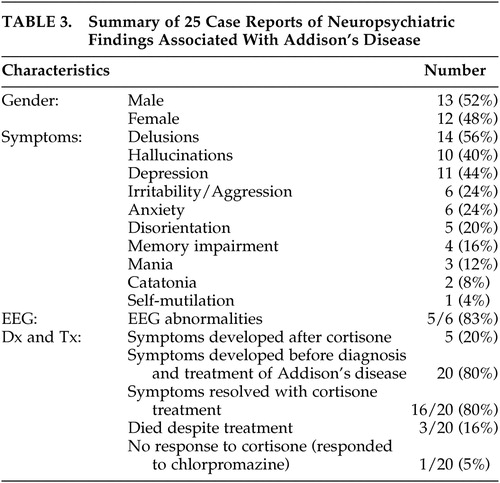
There have been 25 cases 7 – 27 of psychiatric symptoms associated with Addison’s disease reported in the English literature since 1940 ( Table 3 ). Largely these reports have described severe disturbances in functioning and psychosis. Rare presentations of Addison’s disease have included catatonia and self-mutilation. In 80% of the case reports, symptoms occurred before diagnosis and treatment, indicating that the mental status changes are a feature of Addison’s disease and not caused by treatment with cortisone. In the majority of cases, both physical and mental symptoms resolved with cortisone treatment in 1 week. However, mental symptoms occasionally persisted for months and reemerged with the precipitation of an Addisonian crisis.
 |
It is important to note that several terms have been used in the literature to describe the severe mental changes associated with Addison’s disease. These include psychosis, delirium, encephalopathy, and acute organic brain syndrome. Sometimes multiple terms are used to refer to a single patient. The patient we present can also be described by several diagnoses. She clearly had an encephalopathy, as evidenced by her abnormal EEG which showed generalized slowing. She could also be diagnosed with a psychotic disorder due to a general medical condition, namely Addison’s disease, given her prominent delusions and hallucinations that were likely a direct physiological effect of her hypoadrenocortical state. Finally she may have been delirious given her abrupt onset of agitation, disorientation, and perceptual disturbances along with an abnormal EEG. However, this is less likely as she did not have fluctuations in her level of consciousness, was able to focus and sustain attention and did not have any disturbances in speech or language. Regardless of the specific terminology used, it is clear that some patients with Addison’s disease have a disturbance in brain function and may develop a range of neuropsychiatric symptoms as a result.
What Are the Additional Clinical and Laboratory Features That Should Lead One to Consider a Diagnosis of Addison’s Disease?
When Thomas Addison first described the disease in 1855, tuberculosis adrenalitis was the most common cause. Currently, the most common etiology is autoimmune adrenalitis, but infection (including tuberculosis, fungal, HIV and syphilis), metastatic disease, drugs, and adrenoleukodystrophy may also give rise to Addison’s disease. The symptoms and signs observed in Addison’s disease depend on the rate and extent of destruction of the adrenal cortex. 28 The majority of patients with Addison’s disease have gradual destruction of the adrenal gland over months to years. However, bilateral adrenal hemorrhage or infarction or an acute stressor superimposed on chronic adrenal insufficiency can result in fulminant destruction of the adrenal gland and an Addisonian crisis. 28 This can occur in a matter of hours and is often life-threatening.
Patients with chronic adrenal insufficiency may have insidious onset of fatigue, lassitude, malaise, weakness, and weight loss. Hypotension can cause postural dizziness and syncope. Gastrointestinal symptoms including nausea, vomiting, abdominal pain, constipation, and diarrhea are common. Myalgias and arthralgias are frequently observed and rarely flexion contractures of the legs can develop. Decreased libido, thinning of axillary and pubic hair, and amenorrhea may develop. Hyperpigmentation of the skin and mucosal surfaces is the most consistent symptom in Addison’s disease. It is caused by increased action of the melanocyte-stimulating hormone, which is co-secreted with ACTH in response to low circulating cortisol. Vitiligo can occur with autoimmune adrenal insufficiency as a result of destruction of dermal melanocytes.
Electrolyte abnormalities are found in the majority of patients. Hyponatremia is caused by mineralocorticoid deficiency which results in volume contraction and corticosteroid deficiency, leading to an inappropriate secretion of ADH. Patients may report a craving for salt. Hyperkalemia and a mild hyperchloremic metabolic acidosis are caused by mineralocorticoid deficiency. Hypoglycemia can occur after prolonged fasting. Increased blood urea nitrogen and creatinine, a mild normocytic anemia, lymphocytosis, mild eosinophilia and hypercalcemia may be observed.
Patients with Addison’s disease may also present in Addisonian crisis with acute adrenal insufficiency in the setting of a serious infection or other acute stressors. Addisonian crisis is usually precipitated by mineralocorticoid depletion, and patients present with dehydration, hypotension, and shock. Patients may have nonspecific symptoms, including nausea, vomiting, weight loss, abdominal pain, weakness, fatigue, confusion, and even coma. Fever may be caused by an infection, and exaggerated by glucocorticoid deficiency. Patients may have long-standing, undiagnosed Addison’s disease and also present with manifestations of chronic adrenal insufficiency, such as hyperpigmentation and electrolyte abnormalities. Addisonian crisis may also occur after bilateral adrenal hemorrhage or infarction. This should be considered in any patient presenting with severe hypotension or shock, abdominal, flank or back pain, fever, anorexia, nausea or vomiting, confusion, and abdominal rigidity or rebound tenderness in the absence of signs of chronic adrenal insufficiency, such as hyperpigmentation.
A diagnosis of Addison’s disease is based on the measurement of low plasma cortisol, elevated ACTH and the results of a corticotropin stimulation test. In a short corticotropin stimulation test, 250μg of cosyntropin (a synthetic form of ACTH) is given before 10 a.m., and plasma cortisol is measured before the test and 60 minutes after the injection. In patients with Addison’s disease, the adrenal cortex is unable to increase cortisol secretion in response to cosyntropin. When patients present with an Addisonian crisis, glucocorticoid treatment must not be delayed. Blood for serum cortisol, ACTH and serum chemistry should be drawn, and therapy with IV saline and dexamethasone should be initiated immediately. A short corticotropin stimulation test can then be performed as dexamethasone does not interfere with cortisol radioimmunoassay. Following testing, therapy with dexamethasone can be replaced with hydrocortisone. Low aldosterone and high renin are consistent with the diagnosis of Addison’s disease. If the cause of primary adrenal insufficiency is unknown, adrenal autoantibody tests and imaging of the adrenal glands can be performed.
What Is the Association Between Addison’s Disease and Other Endocrine Diseases?
Addison’s disease is most commonly caused by autoimmune destruction of the adrenal cortex, and may be accompanied by other autoimmune endocrine diseases. Autoimmune polyendocrine syndrome type I is an autosomal recessive condition most common in Finns, Sardinians and Iranian Jews that results in mucocutaneous candidiasis, hypoparathyroidism and Addison’s disease. 29 Patients are also at risk of developing type 1 diabetes, hypothyroidism, pernicious anemia, alopecia, vitiligo, hepatitis, ovarian atrophy, and keratitis. Autoimmune polyendocrine syndrome type II is a more common polygenic disorder resulting in a variety of autoimmune conditions. 29 Classically it refers to Addison’s disease plus thyroid autoimmunity (also called Schmidt’s syndrome) or type 1 diabetes, but the definition can be extended to include any two or more organ-specific autoimmune diseases.
Polyendocrine syndromes are believed to result from a failure of self-tolerance to a variety of molecules in different organ tissues. 29 As a result, autoantibodies against self-peptides may form. Greater than 90% of patients with Addison’s disease have autoantibodies to the enzyme 21-hydroxylase, a steroidogenic enzyme present in the adrenal cortex. 29 Specific genetic polymorphisms likely influence which specific autoimmune diseases develop.
In the case presented in this article, the patient was mildly hyperthyroid, with an elevated T3 level. This raises the possibility that thyroid dysregulation may contribute to the development of neuropsychiatric symptoms in patients with Addison’s disease. In the literature, there are four cases that mention thyroid illness in patients with neuropsychiatric symptoms of Addison’s disease. In one case, Hashimoto’s thyroiditis was diagnosed at postmortem. 7 In a second case, there is mention that the patient was treated with “thyroid extract” although no indices are given to indicate that the patient was hypothyroid. 16 In a third case, the patient had a history of treated hypothyroidism and was treated with triiodothyronine in addition to hydrocortisone. 18 The fourth patient had been previously diagnosed with Hashimoto’s thyroiditis. 26 Importantly, there were no active abnormalities in circulating thyroid hormones reported in any of these cases. Therefore, it is unlikely that abnormal levels of thyroid hormones play a significant role in the neuropsychiatric presentation of Addison’s disease.
 |
What Triggered the Addisonian Crisis in This Patient?
Many different stressors can cause acute adrenal insufficiency and trigger an Addisonian crisis. In the patient we describe, several factors likely contributed to her condition. First, 2 weeks prior to her presentation she was unwell and diagnosed with dehydration, food poisoning, and a sinus infection. Second, she was being treated for hypothyroidism and her dose of levothyroxine had recently been increased. Thyroid replacement is felt to increase hepatic corticosteroid metabolism and may precipitate an Addisonian crisis. 29 Third, the patient was undergoing in vitro fertilization. It is possible that the hormone injections she was receiving stimulated an autoimmune process resulting in an autoimmune polyendocrine syndrome. IVF may act in a similar manner to pregnancy, which is known to trigger autoimmune diseases, including postpartum thyroiditis. 29
How Can We Understand the Neuropsychiatric Symptoms Seen in Addison’s Disease?
The pathophysiology of the development of neuropsychiatric symptoms in Addison’s disease remains unclear. Several theories have been proposed:
1) Electrophysiological Abnormalities
EEG recordings are frequently abnormal in Addison’s patients with neuropsychiatric symptoms, indicating that an encephalopathy may develop. Engel and Margolin 3 reported abnormal EEGs in five of eight patients, which commonly showed bursts of 3 to 6 seconds of high voltage potentials with no definite cortical focus. In the 25 cases of neuropsychiatric symptoms associated with Addison’s disease described in the English literature, 7 – 27 EEG results were reported for six patients, 8 , 13 – 15 , 17 , 21 and abnormal in five. The most common abnormality was diffuse slowing. In the case presented in this article, the patient’s initial EEG showed fluctuating slow activity, with 1- to 3-second bursts of slow activity. A repeat EEG 1 week later was markedly abnormal with paroxysmal sharp and slow wave discharges. At 2-month follow-up, her EEG had improved but still showed a sharp and slow wave discharge. The presence of abnormal EEG activity after treatment and clinical resolution of Addison’s disease indicates that there may be persistent brain abnormalities and subtle cognitive impairment.
2) Electrolyte and Metabolic Abnormalities
Hyponatremia occurs in the majority of patients and may contribute to cognitive changes and encephalopathy by causing brain swelling and increased intracranial pressure. Hyponatremic encephalopathy and brain damage occur predominantly in young women who can develop symptomatic hyponatremia at serum sodium values as high as 128 meq/liter. 30 Severe hypoglycemia is occasionally seen in patients with Addison’s disease and can precipitate cognitive changes and even coma. In 1942, Engel and Margolin 3 demonstrated a relationship between hypoglycemia, psychiatric symptoms, and EEG changes. Although hypoglycemia may play a role in some cases, the majority of neuropsychiatric symptoms occur in patients with normal or mildly abnormal blood sugars. It has been suggested that hypoxia secondary to severe hypotension may be responsible for the mental status changes in Addison’s disease. 4 However, cerebral autoregulation is generally capable of compensating for even severe hypotension 31 and as a result hypoxia is not commonly observed in patients with Addison’s disease or even Addisonian crisis.
3) Glucocorticoid Deficiency
The profound decrease in glucocorticoids that underlies many of the manifestations of Addison’s disease is likely to contribute to the development of neuropsychiatric symptoms. Cortisol, the primary glucocorticoid in the body, is lipid soluble and therefore can diffuse through cell membranes and bind to intracellular glucocorticoid receptors in target cells. Once cortisol is bound to its receptor in the cytoplasm, the hormone-receptor complex translocates to the nucleus where it interacts with regulatory DNA sequences and modifies gene transcription. Glucocorticoid receptors are distributed throughout the brain, but are particularly abundant in the hippocampus. It has been demonstrated that adrenalectomy produces massive granule cell death in the dentate gyrus of the hippocampus. 32 – 36 It is possible that granule cell death resulting from cortisol deficiency interrupts the hippocampal tri-synaptic circuit, producing memory impairment and cognitive changes. 37 Recently, glucocorticoids have also been shown to be essential for maintaining prefrontal cortical cognitive function, 38 and likely play other important roles in maintaining a normal mental state. Henkin 39 , 40 – 42 has proposed that a decrease in glucocorticoids results in an increase in neural excitability, leading to enhanced ability to detect sensory input, including taste, olfaction, audition, and proprioception. He has also suggested that decreased glucocorticoids may increase conduction velocity along peripheral axons, while prolonging conduction across synapses. This is felt to change the timing of arrival of signals from the periphery to the CNS, resulting in a loss of perceptual ability and decreased integration of sensory inputs. For example, the ability to recognize the four qualities of taste, understand speech, recognize changes in tone, and localize auditory stimuli is impaired with a decrease in glucocorticoids. It is reasonable to postulate that if patients are receiving abnormally high sensory signals but are unable to process and integrate these signals appropriately, there may be a tendency to develop hallucinations and a lower threshold for psychosis.
4) Increased Endorphins
Increased endorphins may be involved in the etiology of mental changes in Addison’s disease. In response to decreased glucocorticoid production by the adrenal cortex, the anterior pituitary synthesizes a precursor polypeptide, proopiomelanocortin (POMC) which is cleaved to release ACTH and β-endorphin. Therefore, Johnstone et al. 26 proposed that Addison’s disease may be associated with increased endorphins, which could cause psychosis. An association between endorphins and psychosis has been suggested based on the findings of elevated CSF endorphins in schizophrenia patients, 43 the ability of PCP to act on opiate receptors and cause psychosis, 44 and the amelioration of auditory hallucinations with naloxone. 45 As Johnstone et al. 26 indicated, more research is needed to clarify the role of endorphins in producing neuropsychiatric symptoms in Addison’s disease.
5) Associated Adrenoleukodystrophy
Adrenoleukodystrophy (ALD) should be considered in any young man presenting with adrenal insufficiency, particularly when there are associated neuropsychiatric symptoms. ALD is an X-linked peroxisomal disorder characterized by the accumulation of very long chain fatty acids (VLCFA) as a result of defective beta-oxidation. 46 , 47 The disease process preferentially targets the adrenal cortex, gonads and nervous system, resulting in adrenal insufficiency, testicular dysfunction, and central and peripheral demyelination. The disorder classically presents in childhood and is characterized by symptoms of attention deficit disorder, followed by intellectual, behavioral, and neurological deterioration. The illness progresses to a vegetative state within 2 years, followed by death. An adolescent-onset form may present with primary adrenal insufficiency, neurological dysfunction, or psychiatric symptoms. Adult onset ALD comprises different phenotypes, and afflicted individuals may have any combination of adrenal, gonadal, neurological or psychiatric disorders. Presenting features include signs and symptoms of adrenal insufficiency, abnormalities of gait, evidence of upper motor neuron involvement, mania, and psychosis. It is estimated that ADL may account for 10% of all cases of adrenal insufficiency. 48 Of note, up to 60% of young men with ADL may present with adrenal insufficiency before significant neurological dysfunction has developed. 47 , 49 ADL can be distinguished from autoimmune adrenal insufficiency by the absence of circulating adrenal autoantibodies and the presence of elevated levels of VLCFA in serum.
6) Associated Hashimoto Encephalopathy
The patient we describe in this article had a history of treated hypothyroidism and was found at presentation to have extremely elevated antithyroid peroxidase antibodies. Therefore, we suggest that Hashimoto encephalopathy may contribute to the neuropsychiatric symptoms observed in Addison’s disease. Hashimoto encephalopathy is a syndrome characterized by high serum antithyroid antibodies and encephalopathy. 50 Patients are typically hypothyroid but may be euthyroid or hyperthyroid. Clinical features include psychosis (present in 36% of cases), cognitive impairment, stroke-like signs, seizures and myoclonus. Brain CT or MRI is abnormal in 49% of patients, commonly showing white matter abnormalities. Pathological findings include lymphocytic infiltration of blood vessel walls. The treatment of Hashimoto encephalopathy is with glucocorticoids, which are also used to treat Addison’s disease. Autoimmune Addison’s disease frequently occurs with autoimmune thyroid disease as part of an autoimmune polyendocrine syndrome type II. It is possible that a number of patients presenting with neuropsychiatric features associated with Addison’s disease have an undiagnosed Hashimoto encephalopathy that contributes to their symptoms. To our knowledge, this has not been commented on previously in the literature on Addison’s disease or Hashimoto encephalopathy. We suggest it is important to measure serum antithyroid antibodies in addition to thyroid indices in patients presenting with Addison’s disease to determine if there might be an associated Hashimoto encephalopathy.
CONCLUSIONS
In summary, we present a case of a 30-year-old patient referred to psychiatry for psychosis, who was subsequently diagnosed with an Addisonian crisis. Psychiatrists must be aware that an Addisonian crisis can be a fatal emergency that may present with psychiatric symptoms. A review of the literature indicates that disturbances in mood, motivation and behavior occur frequently in Addison’s disease. Psychosis occurs less frequently but is indicative of severe disease and may be the presenting symptom of Addisonian crisis. EEG changes, commonly diffuse slowing and bursts of sharp and slow wave discharges, usually accompany mental status changes. Our case demonstrates that patients may have persistent EEG abnormalities and subtle cognitive changes following treatment and clinical resolution of their Addison’s disease. The etiology of the neuropsychiatric symptoms associated with the disease remains unknown, but may involve electrophysiological, electrolyte and metabolic abnormalities, glucocorticoid deficiency, and increased levels of endorphins. In young men presenting with adrenal insufficiency, adrenoleukodystrophy should be ruled out. In light of our patient’s remarkably high antithyroid antibodies, it is reasonable to postulate that a Hashimoto encephalopathy may also contribute to the neuropsychiatric symptoms associated with Addison’s disease.
1. Addison T: On the Constitutional and Local Effects of Disease of the Suprarenal Capsules. Birmingham, Ala, Classics of Medicine Library, 1980Google Scholar
2. Klippel M: Encephalopathie addisonienne. Rev Neurol 1899; 7:898–899Google Scholar
3. Engel GI, Margolin SG: Neuropsychiatric disturbances in internal disease. Arch Int Med 1942; 70:236–259Google Scholar
4. Sorkin SZ: Addison’s disease. Medicine 1949; 28:371–425Google Scholar
5. Cleghorn RA: Adrenal cortical insufficiency: psychological and neurological observations. Can Med Assoc J 1951; 65:449–454Google Scholar
6. Smith CK, Barish J, Correa J, et al: Psychiatric disturbance in endocrinologic disease. Psychosom Med 1972; 34:69–86Google Scholar
7. Rushton JG, Cragg RW, Stalker LK: Spontaneous hypoglycemia due to atrophy of the adrenal glands. Arch Int Med 1940; 66:531–540Google Scholar
8. Engel GL, Margolin SG: Neuropsychiatric disturbance in Addison’s disease and role of impaired carbohydrate metabolism in production of abnormal cerebral function. Arch Neurol Psychiatry 1941; 45:881–884Google Scholar
9. Gorman WF: Psychosis in Addison’s disease. Dis Nerv Sys 1947; 9:7–71Google Scholar
10. Craddock WL, Zeller NH: Use of ECT in a case of Addison’s disease. Arch Int Med 1952; 90:392–394Google Scholar
11. Cleghorn RA, Pattee CJ: Psychologic changes in 3 cases of Addison’s disease during treatment with cortisone. J Clin Endocrinol Metab 1954; 14:344–352Google Scholar
12. Cumming J, Kort K: Apparent reversal by cortisone of an electroconvulsive refractory state in a psychotic patient with Addison’s disease. Can Med Assoc J 1956; 74:291–292Google Scholar
13. Smith CM: Paranoid behavior in Addison’s disease: report of a case. Can Psychiatr Assoc J 1958; 3:145–154Google Scholar
14. Wolff HD, Huston PE: Schizophrenia associated with Addison’s disease. Am J Psychiatry 1959; 116:365–367Google Scholar
15. Cohen SI, Marks IM: Prolonged organic psychosis with recovery in Addison’s disease. J Neurol Neurosurg Psychiatry 1961; 24:366–368Google Scholar
16. Mcculloch TAH, Calverley MO: Addison’s Disease with psychosis. Can Med Assoc J 1961; 85:31–33Google Scholar
17. Mcfarland HR: Addison’s disease and related psychoses. Compr Psychiatry 1963; 4:90–95Google Scholar
18. Harper MA, Earnshaw BA: Combined adrenal and thyroid deficiency (Schmidt’s Syndrome) presenting as an acute psychosis. Med J Aust 1970; 1:546–548Google Scholar
19. Bugiani O, Balestreri R: Addison-Schilder’s disease: report of a case with probable relapsing addisonian encephalopathy. Confin Neurol 1971; 33:208–220Google Scholar
20. Mattsson B: Addison’s disease and psychosis. Acta Psychiatr Scand 1974; 255(suppl):203–210Google Scholar
21. Lever EG, Stansfeld SA: Addison’s disease, psychosis, and the syndrome of inappropriate secretion of ADH. Br J Psychiatry 1983; 143:406–410Google Scholar
22. Rajathurai A, Chazan BI, Jeans JE: Self-mutilation as a feature of Addison’s disease. Br Med J 1983; 287:1027Google Scholar
23. Demilio L, Dackis CA, Gold MS, et al: Addison’s disease initially diagnosed as bereavement and conversion disorder. Am J Psychiatry 1984; 141:1647–1648Google Scholar
24. Varadaraj R, Cooper AJ: Addison’s disease presenting with psychiatric symptoms. Am J Psychiatry 1986; 143:553–554Google Scholar
25. Malu AO, Sanusi BR, Obineche EN: Addison’s disease presenting with marked eosinophilia and psychosis. Trop Geogr Med 1988; 40:241–243Google Scholar
26. Johnstone PA, Rundell JR, Esposito M: Mental status changes of Addison’s disease. Psychosomatics 1990; 31:103–107Google Scholar
27. Thompson WF: Psychiatric aspects of Addison’s disease: report of a case. Medical Annals of the District of Columbia 1973; 42:62–64Google Scholar
28. Oelkers W: Adrenal Insufficiency. N Engl J Med 1996; 335:1206–1212Google Scholar
29. Eisenbarth GS, Gottlieb PA: Autoimmune polyendocrine syndromes. N Engl J Med 2004; 350: 2068–2079Google Scholar
30. Moritz ML, Ayus CJ: The pathophysiology and treatment of hyponatraemic encephalopathy: an update. Nephrol Dial Transplant 2003; 18:2486–2491Google Scholar
31. Michiels C: Physiological and pathological responses to hypoxia. Am J Path 2004; 164:1875–1882Google Scholar
32. Sloviter RS, Valiquette G, Abrams GM, et al: Selective loss of hippocampal granule cells in the mature rat brain after adrenalectomy. Science 1989; 243:535–538Google Scholar
33. Gould E, Woolley CS, McEwen BS: Short-term glucocorticoid manipulations affect neuronal morphology and survival in the adult dentate gyrus. Neuroscience 1990; 37:367–375Google Scholar
34. Gould E, Cameron HA, Daniels DC, et al: Adrenal hormones suppress cell division in the adult rat dentate gyrus. J Neurosci 1992; 12:3642–3650Google Scholar
35. McNeill TH, Masters JN, Finch CE: Effect of chronic adrenalectomy on neuron loss and distribution of sulfated glycoprotein-2 in the dentate gyrus of prepubertal rats. Exp Neurol 1991; 111:140–144Google Scholar
36. Sloviter RS, Sollas AL, Dean E, et al: Adrenalectomy-induced granule cell degeneration in the rat hippocampal dentate gyrus: characterization of an in vivo model of controlled neuronal death. J Comp Neurol 1993; 330:324–336Google Scholar
37. Squire LR, Zola-Morgan S: Memory: brain systems and behavior. Trends Neurosci 1988; 11:170–175Google Scholar
38. Mizoguchi K, Ishige A, Takeda S, et al: Endogenous glucocorticoids are essential for maintaining prefrontal cortical cognitive function. J Neurosci 2004; 24:5492–5499Google Scholar
39. Henkin RI: The effects of corticosteroids and ACTH on sensory systems. Prog Brain Res 1970; 32:270–294Google Scholar
40. Henkin RU, Gill JR, Warmolts JR, et al: Steroid-dependent increase of nerve conduction velocity in adrenal insufficiency. J Clin Invest 1963; 42:941Google Scholar
41. Henkin RI, Gill JR, Bartter FC: Studies on taste thresholds in normal man and in patients with adrenal cortical insufficiency: the role of adrenal cortical steroids and of serum sodium concentration. J Clin Invest 1973; 42:727–735Google Scholar
42. Henkin FI, Daly RL: Auditory detection and perception in normal man and in patients with adrenal cortical insufficiency: effect of adrenal cortical steroids. J Clin Invest 1968; 47:1269–1280Google Scholar
43. Lindstrom LH, Widerlov E, Gunne LM, et al: CSF: clinical correlations to some psychotic states. Acta Psychiatr Scand 1978; 57:153–164Google Scholar
44. Steinpresis RE: The behavioral and neurochemical effects of phencyclidine in humans and animals: some implications for modeling psychosis. Behav Brain Res 1996; 74:45–55Google Scholar
45. Lehmann H, Nair NPV, Kline NS: Beta-endorphin and naloxone in psychiatric patients: clinical and biological effects. Am J Psychiatry 1979; 136:762–766Google Scholar
46. Garside S, Rosebush PI, Levinson AJ, et al: Late-onset adrenoleukodystrophy associated with long-standing psychiatric symptoms. J Clin Psychiatry 1999; 60:460–468Google Scholar
47. Rosebush PI, Garside S, Levinson AJ, et al: The neuropsychiatry of adult-onset adrenoleukodystrophy. J Neuropsychiatry Clin Neurosci 1999; 11:315–327Google Scholar
48. Laureti S, Aubourg P, Calcinaro F, et al: Etiological diagnosis of primary adrenal insufficiency using an original flow chart of immune and biochemical markers. J Clin Endocrinol Metab 1998; 83:3163–3168Google Scholar
49. Sadeghi-Dejad A, Senior B: Adrenomyeloneuropathy presenting as Addison’s disease in childhood. N Engl J Med 1990; 322:13–16Google Scholar
50. Chong JY, Rowland LP, Utiger RD: Hashimoto encephalopathy. Arch Gen Neurol 2003; 60:164–171Google Scholar

