
Considering the Role of the Amygdala in Psychotic Illness
Abstract
For many years, the structures of the medial temporal lobe have been implicated in the pathogenesis of schizophrenia. Recent hypotheses, based on data from MRI and functional imaging, propose that disruption of frontotemporal neural networks may be an anatomical substrate of schizophrenia. Many studies have focused on possible abnormalities of the hippocampus within this network. However, the role of the amygdala has been little studied because of the relative complexity of its structure and the paucity of patients with confined amygdaloid lesions. The authors present a case of chronic psychosis in which postmortem findings reveal lesions in and adjacent to the left amygdala. They use this case to review what is known of the functional anatomy of the amygdala and its possible role in some psychoses.
For the last 20 years, the role of the limbic system in psychotic illness has been a matter of much debate. Early electroencephalographic studies of schizophrenic patients suggested connections between the limbic structures of the medial temporal lobe and the ventral striatum as a possible anatomic substrate of schizophrenia.1,2 The striking phenomenological similarities between psychotic patients and patients with temporal lobe epilepsy were cited, along with EEG evidence of temporal lobe abnormalities in psychotic patients. Furthermore, it was recognized that the amygdala has androgen receptors, a potential mediator of symptom onset during adolescence.2 With the advent of MRI, volumetric studies documented changes in medial temporal lobe structures between schizophrenic patients and control subjects.3,4
In the last decade, with the increased delineation of the negative symptoms of schizophrenia and advances in imaging technology, there has been a shift in focus toward brain regions underlying the cognitive deficits in this illness. Consequently, cortical areas such as the dorsolateral prefrontal cortex, which mediate many of these cognitive functions, have been studied intensely.5,6 Most recently, several schizophrenia studies have linked functional abnormalities in prefrontal cortex, as measured by PET, to pathology in the medial temporal lobe, as measured by MRI. These data have been used to build hypotheses about the neural networks that may underlie these two areas.7,8
Despite recent interest in frontotemporal networks in schizophrenia, there are few studies of the amygdala, a major component of the limbic system that is located in this region. Although Stevens2 suggested that the amygdala plays a role in positive symptoms (before that term was in use), few data from clinical settings have been collected to support this view. We present a case report of a young woman who had a deteriorating psychotic illness and whose autopsy findings included lesions in the left amygdala. These findings are discussed in the context of the neurodevelopmental hypothesis of schizophrenia and with respect to the neuroanatomy and physiology of the amygdala.
CASE REPORT
Ms. A. was first admitted to the psychiatric unit of Strong Memorial Hospital, Rochester, NY, in autumn of 1979 at the age of 20. By report of her mother, she was the product of a normal gestation and normal spontaneous vaginal delivery and weighed 7 pounds at birth. There was no childhood history of high fever, seizure, or serious illness. The patient's psychiatric and medical history were unremarkable until age 15, when she sustained head trauma secondary to being kicked by a horse. There was no loss of consciousness or apparent neurological sequela related to this event. Ms. A. began considering herself a “born-again” Christian while in high school, but she did not increase her religious activity at that time. At age 18, she enrolled in the state university. She began having vivid hallucinations at that time, although these were only reported retrospectively. Her parents became concerned about her affiliation with a cult-like church. During the second semester of her freshman year, Ms. A. dropped out of college because of her fears of the occult, involving, in particular, the idea that a professor was trying to hypnotize her. At home, she had delusions of influence, accusing her father of hypnotizing her. She described unusual thoughts to her mother—for example, the ideas that demons were in her bedroom and that acquaintances were controlling her. Although the patient was employed during this time, she was hyperreligious, reciting the Bible frequently and sending money to a faith-healing church.
In summer 1979, slightly more than one year after dropping out of college, Ms. A. poured boiling water on her leg, saying she was being compelled to do this by forces outside her control, and a week later she bought “health food” for her leg. Two months afterward she was admitted to a local hospital and evaluated for a week. Her diagnosis at that time was chronic paranoid schizophrenia, and she was begun on low-dose trifluoperazine. Nine days after discharge, Ms. A. presented to the emergency department of another hospital with apparent disorientation, confusion, and fluctuating levels of consciousness. The patient uttered repetitive sentences, such as “Wake me up,” and held the cross she wore up in the air. During the ensuing months she continued to hallucinate. She stopped her medication a month after the hospital evaluation. Two months later, Ms. A. amputated the distal third of her tongue in response to commands from inner voices. She said she had felt “overcome” by “demonic forces” and “forced downstairs by air cushions.” Using a pillow to stop the bleeding, she called 911 emergency services. After surgical reattachment of the tongue fragment, the patient was admitted to the Department of Psychiatry at Strong Memorial Hospital for 3 months. While hospitalized, she revealed complex violent sexual hallucinations (including visual hallucinations of being “raped by Prince Charles”) in addition to other symptomatology. It was noted that psychosis seemed to be exacerbated perimenstrually.
Results of physical and neurological examinations conducted during this hospital stay were unremarkable. Lumbar puncture was performed, and cerebral spinal fluid studies for glucose, protein, rapid plasma reagin (RPR), viral titers, bacterial cultures, and immunoglobulin G were all normal. Neuropsychological testing was within normal limits. A positive serum RPR with negative fluorescent treponemal antibody test prompted autoimmune workup. Autoimmune studies were as follows: initial antinuclear antibody test (ANA) positive at 1:1,000 dilution, speckled pattern, elevated hemolytic complement (CH50) of 114, and elevated immunoglobulin M (462%) on electrophoresis. Cryoglobulins, Raye cell test, C1q, rheumatoid factor, and skin biopsy for systemic lupus erythematosus were all negative. Serial erythrocyte sedimentation rates were 43, 16, 25, and 10. A repeat ANA 1 week after the initial result was trace positive at 1:1,000 dilution. Clinically, the patient did not meet criteria for systemic lupus erythematosus or any other autoimmune disorder. The differential diagnosis was “atypical psychosis” versus temporal lobe epilepsy. Sequential, sustained trials with adequate doses of carbamazepine, haloperidol, chlorpromazine, and molindone were not successful.
Over the next 13 years, Ms. A. had eight acute inpatient hospitalizations and three lengthy admissions on the teaching service at the local state hospital (totaling 79 months) for exacerbations of psychosis and suicidal ideations, often bizarre in nature. In addition to the medications listed above, she received thorough therapeutic trials of diazepam, flurazepam, lithium, valproic acid, phenelzine, thiothixene, phenytoin, lorazepam, clonazepam, thioridazine, amitriptyline/perphenazine (Triavil), perphenazine, fluphenazine, fluoxetine, and bupropion, with little success. Numerous psychotherapeutic interventions were tried, including insight-oriented psychotherapy, amobarbital interview and hypnosis, behavioral modification, supportive therapy with a psychoeducational focus, family therapy, and various supportive residential and occupational placements. In total, 16 EEG studies were done; 4 revealed paroxysmal bilateral spike and wave activity, and the rest were within normal limits. CT and MRI studies performed during this period inconsistently showed temporal asymmetry (R>L).
Ms. A.'s only persistent medical issue was periodic flares of genital herpes. Five years after her initial psychiatric diagnosis, she was noted to have a fluctuating neutropenia not associated with medication (white blood count 2,100–8,400) and mild anemia (hematocrit ranging from 34 to >40). Thyroid functions were normal until 1992, when thyroid-stimulating hormone was slightly elevated to 11.08, with a subsequent normal value 2 weeks later. ANA titers were often normal and occasionally weakly positive (1:160).
Although Ms. A. suffered a chronically deteriorating clinical course, with persistent hallucinations and the belief that she was being controlled or hypnotized, she was noted to retain much of her affective tone, and she engaged with staff and fellow patients. Her thought processing was generally lucid, although there was formal thought disorder during periods of psychotic exacerbation. Repeated neuropsychologic testing showed a general decline in intellectual function. Increasingly poor performance on tests of concentration and attention made it difficult to interpret tests of other functions such as verbal fluency and short-term memory.
Ms. A.'s discharge from her last psychiatric hospitalization was in late 1992. She was taking lithium 900 mg qd and haloperidol 15 mg qd in divided doses, and fluoxetine 20 mg qd. During this hospitalization, she had received a diagnosis of mild sleep apnea. Two weeks later, the patient developed a sore throat, confusion, and slurred speech. After several evaluations in the emergency department, she was admitted to the Medicine Service of Strong Memorial Hospital with stridorous breathing, progressive hyponatremia (sodium values declining from 135 to 130 to 123 in the three days prior to admission), and complaints of chest pain. She had remained on the above medications, and her lithium level was 0.46 two days prior to admission. She expired precipitously early on the morning of the day after admission. No cause of death was established, despite an autopsy and toxicological tests (Office of the Medical Examiner, Monroe County, NY) and case review by the faculty of the Department of Medicine, by outside consultants, and by the State Department of Health.
Pathologic findings. The postmortem examination (autopsy) did not reveal an anatomic cause of death. From the available clinical, laboratory, and pathologic data, it appeared that the patient died of a toxic-metabolic process in which drug therapy, hyponatremia, and hypothyroidism may have played some role. The systemic autopsy findings were confined to some acute lung and gastrointestinal tract congestion and thyroid lesions. The thyroid gland showed lobulations due to fibrosis and infiltration by lymphocytes, with Hürthle cell metaplasia of follicular epithelium. There was also a small focus of papillary carcinoma. The pituitary was enlarged as a result of thyrotroph cell hyperplasia. Small numbers of lymphocytes were also found in the pericardium. These findings were consistent with an active systemic infection, perhaps viral, or may have reflected some immune reaction. However, neither infection nor immune reaction could be established, and such diagnostic possibilities remain speculative.
Of more specific interest were those lesions found in the central nervous system. The brain demonstrated diffuse, mild cerebral edema with some uncal grooving. Microscopically, mild intramyelinic edema was observed in the myelinated fibers of pons, internal capsule, and frontal white matter. These alterations were consistent with an acute toxic-metabolic process in which fluid and, probably, small ions accumulate between the myelin lamellae because of osmotic shifts; such a process has been reported in central pontine myelinolysis.9 There was also a mild and chronic lymphocytic perivasculitis of the cingulate, left occipital and right superior posterior temporal cortex, left amygdala, pons, and posterior pituitary. Neither demyelination nor necrosis was associated with this perivasculitis. One could speculate that these findings reflect a chronic infectious process that could have played a role in the patient's illness. Alternatively, they may be more consistent with an epiphenomenon in the form of an acute viral infection or active immunologic response to an unknown, noninfectious antigen. Such inflammatory infiltrates are occasionally seen at autopsy in patients without neurologic or psychiatric disease.
The most noteworthy lesion with respect to the patient's psychiatric illness, however, was found in the left amygdala region. There were no specific asymmetries appreciated grossly in the temporal lobe. Microscopically, however, there was a focal decrease in myelinated fibers and replacement by fibrillary astrogliosis (scarring) with corpora amylacea in the left temporal white matter (Figure 1) adjacent to the basolateral portion of the amygdaloid complex.
In addition, within the basolateral nucleus of the amygdala, there were two small hamartias, or regions of microdysgenesis. In these areas, primitive or germinal cells were admixed with large pyramidal cells. The hamartias consisted of clusters of round nuclei, often with a prominent hematoxylinophilic nucleolus, surrounded by a perinuclear clear space (Figure 2). The hamartias were so small that they could be seen in only a few serial sections. The Luxol fast blue PAS–stained section was decolorized and immunostained with an antibody to bcl-2, a marker of immature developing neurons (processed by Dr. Anthony Yachnis, University of Florida).10 The cells did not label, but this was interpreted as a false negative stain. Positive immunostaining is obtained only with freshly cut sections (A. T. Yachnis, personal communication). This particular section had been stained and stored for 3 years before we consulted Dr. Yachnis regarding this technique. The specificity of the hamartias was confirmed by their absence in the corresponding levels of the contralateral (right) amygdala and in an additional 20 control amygdalas at the same level.
Within the area of the hamartias, there also was a single swollen eosinophilic structure, which most likely represents a neuraxonal spheroid or swelling (Figure 2). Spheroids most commonly occur following brain trauma, in which they have been referred to as shearing injuries.11 This structure was examined ultrastructurally by using the pop-off technique.12 Ultrastructural preservation was understandably poor but revealed an accumulation of fairly uniform structures most consistent with residual lysosomal autophagic bodies (Figure 3). Such reactive axonal spheroids are consistent with remote destructive processes such as trauma.
The remaining sections of the central nervous system were essentially unremarkable except for a small cortical hamartia in layer 3 of the right occipital cortex. This lesion consisted of a cluster of ill-defined neuroglial cells that also was so small that it was lost in serial sections and could not be studied further.
SUMMARY OF FINDINGS
The above case is provocative on a number of levels, not the least of which is the patient's relentless course of florid psychosis (positive symptoms) and her abrupt demise. The cause of the latter has never been clarified, although a metabolic imbalance seems most likely. Efforts during the patient's life to understand her disease in terms of biological, psychological, and social paradigms did not yield a definitive diagnosis or particularly efficacious treatment interventions. Her progressive course was associated with deterioration in her ability to work and live independently. She eventually manifested typical “negative symptoms” and had difficulties with motivation (for example, on her last admission she stated repeatedly that she wanted to “get on with life” now that “the voices were better,” but she stayed in bed much of the day) and with anergia and inattention. In the face of closely monitored therapeutic trials of medication, the patient had multiple, recurrent exacerbations of psychosis requiring hospitalization. Ms A. remained well related socially, and she often discussed her dislike of taking psychotropics, saying they made her feel tired and “decrepit.” Although she remained delusional, she spoke hopefully with staff on her last psychiatric hospitalization of her wish to be better and to be able to marry and have a family.
Although several of Ms. A.'s early psychotic presentations were reminiscent of partial complex epilepsy, associated with sudden onset, aura of fear, confusion, and complex automatisms, the course of her illness was atypical for this diagnosis. We later learned that these events occurred several years after the onset of Ms. A.'s other psychotic symptoms. Later psychotic exacerbations did not follow this pattern of acute onset with apparent automatisms and acute confusion. Electroencephalographic studies captured left-sided spike and wave discharges inconsistently. Although EEG studies done when the patient was on benzodiazepines or carbamazepine were normal, she remained unchanged symptomatically. Prolonged therapeutic trials of anticonvulsant medication also failed to improve Ms. A.'s level of function or reduce her need for hospitalization. As well, there was never any history of grand mal seizure, indicative of secondary generalization. Neuroimaging procedures indicated a possible asymmetry of the temporal horns, but the clinical relevance of this was unclear.
DISCUSSION
The case of Ms. A. provides an unusual opportunity to consider the role of amygdaloid pathology in the genesis of some psychotic states. The amygdala is located in the temporal lobe, just rostral to the hippocampus. The amygdala is not a homogeneous structure; it is composed of several nuclear groups with an array of interconnections between cortex, basal ganglia, hippocampus, thalamus, brainstem, and hypothalamus. It is generally accepted that the amygdala plays a role in attaching emotional significance to environmental stimuli.13–15 For example, animal studies show that amygdaloid neurons fire preferentially when stimuli that are species-specific are presented (e.g., when a cat is presented to a rat).16,17 With respect to primates, including humans, there is accumulating evidence from lesion and stimulation studies that the amygdala determines the emotional valence of a stimulus. Thus, dysfunction of the amygdala may result in disruption of both the type and the amount of emotional significance that is assigned to a given environmental cue.
Lesion Studies
Historically, lesion studies have provided one important means for understanding the functional significance of specific brain regions. With respect to the amygdala, studies of amygdalectomy in adult monkeys have demonstrated a lack of fear in response to relevant environmental stimuli, as well as an inappropriate tendency to explore all stimuli (hypermetamorphosis), known as the Klüver-Bucy syndrome.18–20 More recently, studies of amygdalectomy in infant monkeys demonstrate that the Klüver-Bucy syndrome is not seen when the amygdala is lesioned early in brain development. In these studies, monkeys had deficits in social behaviors, such as active withdrawal from normal peers and greater passivity.21 These results raise the important issue of brain development as a variable in the behavioral expression of a “deficit lesion.”
In humans, studies of amygdalectomy are limited by lack of appropriate controls and great variability in the size of surgical lesions. Overall, there is a general decrease in emotional output (in particular, aggressive behavior) and a change in the ability to recognize facial expressions in amygdalectomized patients.22 Recently, Adolphs et al.23 reported an unusual case of an adult patient with bilateral amygdala lesions due to calcium depositions resulting from Urbach-Wiethe disease, a rare condition. This patient was neither psychotic nor depressed, but was unable to recognize fear among various facial expressions. Furthermore, she was unable to recognize more than one emotion in a single facial expression. The authors propose that these findings provide evidence for a specific cognitive role of the amygdala in affective recognition and that they also challenge general notions regarding the amygdala's involvement in emotion.
Stimulation Studies
Stimulation of the amygdala results in clinical phenomena that contrast strikingly with the picture of a deficit lesion, providing insights into the behavioral effects of neuronal firing (rather than lack of firing) in this region. Early stimulation studies of the amygdala in awake humans elicited a range of emotional and perceptual states, including fear and anxiety, complex hallucinations, and feelings of familiarity (déjà vu).24–26 Halgren et al.24 noted that the evocation of these effects depended on the presence of after-depolarizations, an indication that a discharge had propagated to other brain regions. In addition, the content of the effect (for example, a hallucination) depended on the patient's personality and past experiences, rather than the precise region stimulated. For example, a patient might hallucinate a voice that would be identified as that of a family member. In general, stimulation studies in awake humans have been limited to patient populations with preexisting brain disease, usually temporal lobe epilepsy (TLE) and schizophrenia. Nonetheless, these studies are notable for their evocation of symptoms that are remarkably like the positive symptoms of schizophrenia.
Temporal Lobe Epilepsy and Psychosis
Temporal lobe epilepsy is an endogenous model of abnormal stimulation of structures of the medial temporal lobe, including the amygdala. Patients with TLE exhibit a range of psychomotor behaviors during the ictal phase, including affective changes (usually toward negative affects such as fear), visual and auditory phenomena that take the form of an actual experience, déjà vu, and automatisms. These phenomena can coalesce, creating an intense sense of reality. Pleasurable emotional changes have been reported in psychomotor seizures but are less common than negative states, specifically fear. Furthermore, psychosexual phenomena—of the type reported by Ms. A.—have been reported, but only in female patients. This has led to speculation regarding gender differences in brain lateralization of sexual experience.27
Psychosis may be observed ictally, interictally, and also as an enduring feature of TLE. Interictal psychosis in TLE patients was first described by Hill28 and Pond29 in the 1950s. Both groups noted that some individuals developed chronic interictal psychoses as the illness progressed, associated with characteristic systematized delusions, ideas of reference, and auditory hallucinations. However, they distinguished these psychoses from schizophrenia in that patients retained “a sense of affective warmth.”29 Slater and Beard, who coined the term schizophrenia-like psychosis, characterized a subgroup of TLE patients with psychosis as having delusions, often of a religious nature, as well as prominent ideation of being controlled or influenced. Hallucinations tended to be complex and deeply meaningful.30 (Slater and Beard's description is particularly reminiscent of the clinical picture seen in the case of Ms. A.) Since these early descriptive studies, epileptic psychoses have been increasingly examined in controlled studies to explore the relationship between psychosis, seizure phenomena, and associated neuropathology.31–35
Role of the Amygdala in TLE
Both the amygdala and the hippocampus are deep limbic structures that are highly epileptogenic and are associated with TLE. Historically, the hippocampus has received more attention as a focus for TLE, perhaps unduly. In humans, the amygdala may be overlooked as a seizure focus owing to intrinsic characteristics of the electrical field it generates.27 Because of the less organized cellular and chemical structure of the amygdala, electrical fields created by discharging amygdaloid neurons are unlikely to conduct outside the structure itself and are therefore unlikely to be captured by surface EEG. Therefore, the incidence of abnormal discharge from the amygdala may be underestimated.
Furthermore, the amygdala has shown extreme vulnerability to kindling. Kindling was first noticed as a “side effect” of stimulation studies in animals: a stimulation that in itself does not provoke a seizure would do so if repeated often enough. Thus, the “seizure threshold” of the original stimulation site was lowered by repeated impulses. In addition, once an afterdischarge was generated, there often was propagation to distal sites, with “transfer” of kindling to other brain regions. Thus, kindling can effect changes in synapses at great distances from the originally stimulated site. Therefore, kindling may explain how abnormal stimulation of vulnerable regions can cause plastic changes in brain sites “downstream” from the original stimulation site. Kindling is a possible mechanism by which abnormal firing in deep limbic structures may result in psychotic symptoms years after seizure onset.36,37
Clinical Correlation
Ms. A.'s illness was atypical for TLE; however, several phenomenologic features are consistent with overactivation, rather than underactivation, of the amygdaloid focus. We propose that the case of Ms. A. provides a complementary perspective to the “deficit lesion” model of amygdaloid dysfunction—suggesting a pattern in which dysregulated, abnormal firing occurs in this part of the brain. Whereas a deficit lesion of the amygdala may result in mostly cognitive dysfunction (as in the case of Adolphs et al.23), abnormal firing may cause perceptual disturbance and hallucinations acutely. Eventually, such symptoms may become “hard wired” via a kindling mechanism. In the case of Ms. A., complex hallucinations had intense affective components involving sexual, violent, and romantic content. Unlike Adolphs's patient, who missed affective cues, Ms. A. had an overly referential quality to her thinking, leading her to over-read rather than under-read affective cues in the world around her. She often experienced inappropriate, overwhelming emotions, particularly fear and paranoia. In this regard, Ms. A. attributed too much personal significance to phenomena in her environment. As is typical in paranoid ideation, she interpreted unrelated environmental cues as having special reference to her.
Ms. A. had neither clinical nor electrophysiologic evidence of TLE. As well, her clinical picture was not fully consonant with schizophrenia. Nonetheless, the clinical course of her illness had elements of both idiopathic schizophrenia and TLE, both in its debilitating course and in its periodic exacerbations. At the time of onset in late adolescence, Ms. A. was a high-functioning individual, even though her hyperreligiosity may have been an early sign of impending psychosis. By age 18, the patient was having visual hallucinations and ideas of reference, which led to her dropping out of college within 6 months. After 1 year of untreated psychosis, she exhibited episodes of complex hallucinations (many with sexual content), automatisms, high anxiety, and confusion, which precipitated her early hospitalizations. Over the next decade, she developed a more persistent delusional system and negative symptomatology. With respect to EEG findings, the localization of limbic epileptic foci by surface recording is highly variable. Because sporadically captured EEG abnormalities were poorly localized, it must remain undetermined whether they represented poorly captured amygdaloid discharge. Despite the lack of definitive clinical or EEG confirmation, a kindling model of amygdaloid pathophysiology may explain both the course and the form of her illness better than a diagnostic category could.
Neuropathologic Correlates of TLE and Schizophrenia
Neuropathologic studies of TLE resections have examined the relationship of abnormalities in the medial temporal lobe, including the amygdala, to psychotic symptomatology. Microscopic hamartias, such as were identified in the case of Ms. A., have been identified in about 15% of temporal resections for intractable epilepsy.38 Among patients resected for TLE, psychotic symptoms are disproportionately high in patients with developmental, rather than destructive, lesions of the temporal lobe, including hamartias.34,39–41 Immunocytochemical techniques now provide a way to investigate the developmental origins of hamartias. A protein expressed in brain tissue, bcl-2, is hypothesized to function as a regulator of programmed cell death in early brain development;42 therefore, it is considered a marker of developing neurons. Yachnis et al.10 found that in normal fetal brain, bcl-2 was expressed strongly in postmitotic, migrating neurons between 6 and 20 weeks of gestational age, after which it decreased sharply. Within temporal lobe sections from adult TLE patients, Yachnis et al.10 showed that bcl-2 is persistently and aberrantly expressed within glioneuronal hamartias of the type found in the brain of Ms. A.
A recent whole-brain study of epileptics with and without interictal psychosis showed that three features distinguished the psychosis group: enlarged ventricles, periventricular gliosis, and an excess of acquired focal lesions.35 Interestingly, all of these findings have been reported in postmortem studies of “pure” schizophrenia.43–46 In the case of Ms. A., astrogliosis of the temporal lobe, as well as an “acquired focal lesion” in the form of the spheroid, were features of her brain pathology. With respect to “pure” schizophrenia, postmortem studies comparing schizophrenic brains with controls have documented dystrophic changes in widespread cortical and subcortical regions.47,48 However, there is a paucity of data on the amygdala. Only one study, to our knowledge, has examined the amygdala in schizophrenic subjects; there was no decrease in basolateral nuclear group (BLNG) cell counts in schizophrenic brains compared with controls.49
With respect to the relationship between psychosis and abnormal neuronal discharge, several questions emerge. One is whether in certain cases psychosis eventually results from aberrantly firing foci, as in a kindling model. Another is whether in some schizophrenic phenotypes abnormal firing (seizure or seizure-like phenomena) is but one early manifestation of disturbed synaptogenesis. “Wiring” abnormalities that eventually give rise to schizophrenia may also be epileptogenic early in the course for some individuals, particularly if lesions are severe enough or occur in vulnerable neuronal populations.
Neuropathologic Correlates
The neuropathologic findings in the case of Ms. A. are subtle, but they do correlate well with the clinical phenomenology and what is known of the anatomy and development of this brain region. The stainability of hamartias identical to hers with antibodies to bcl-2 is evidence that there is an element of developmental arrest in the left basolateral amygdala, possibly at 6 to 20 weeks of gestational age. Another area of microdysgenesis in layer 3 of the right occipital cortex further supports the notion of a global malformative process, with regions of faulty or incomplete wiring. The relationship of these dysgenetic changes to subtle findings of an “acquired” brain lesion (decreased myelin staining, chronic astrogliosis, and the neuraxonal spheroid) remains unclear. However, one can speculate that there was some in utero insult to this area or trauma to the left temporal lobe at birth (a benign or subclinical variant of incisural sclerosis50), which might have interfered with the maturation of nearby vulnerable cells in the region. On the other hand, the myelin loss, scarring, and spheroid might have resulted from later trauma, perhaps at age 15 when the patient sustained head trauma. Perhaps this trauma, either alone or in concert with the hamartia, produced her clinical symptomatology. Although the hamartia appears to be datable to the in utero period, we cannot specifically date the loss of myelinated fibers, spheroid, or fibrillary gliosis except to say that they are chronic and of at least several years' duration.
Anatomic Features of the Basolateral Nuclear Group
Ms. A.'s amygdaloid lesions were primarily within or adjacent to the BLNG of the left amygdala (see schematic in Figure 4). As Alheid and Heimer51 point out, animal studies reveal that the BLNG is a “quasi-cortical” structure that is distinct in specific ways from the other amygdaloid nuclei. Although the BLNG lacks the laminar structure of cortex and lies deep in the brain, it shares a number of similarities with cortex.51 The cells have a pyramidal morphology. Like cortex, and unlike other nuclei of the amygdala, the BLNG contains glutamate, an excitatory neurotransmitter, as its main transmitter.52,53 In primates, the BLNG has direct projections to limbic-related cortex as well as direct projections to limbic-related ventral striatum and basal forebrain.54–57 Thus, the BLNG provides excitatory input to limbic cortex, in particular orbital and insular cortices, as well as to its subcortical targets. Recently, extensive neuronal tract tracing studies in primates have elucidated the “anterior limbic-prefrontal network.”57 These experiments demonstrate that projections from the basolateral amygdala to the lateral, posterior, and medial orbital cortex are a major portion of the limbic-prefrontal network.57 Regions of limbic-associated cortex that receive amygdaloid input from the BLNG project back to the ventral striatum.58,59
The nucleus accumbens has long been hypothesized to be the anatomical substrate mediating psychotic symptoms. More recent data show that the entire rostroventral area of the striatum, which includes the nucleus accumbens, is considered limbic-related. This region is referred to as the ventral striatum. Besides receiving excitatory input from limbic-related cortex and the amygdala, the ventral striatum is modulated by the dopaminergic neurons of the ventral midbrain.60 For many years, the proposed mechanism by which conventional antipsychotic drugs work has been blockade of dopamine transmission in this region.61 In particular, the part of the ventral striatum known as the shell of the nucleus accumbens is a likely site for the action of antipsychotic drugs.62,63 This region receives a dense projection from the BLNG. Excitatory inputs from the BLNG may act as a “gate,” shifting ventral striatal neurons into a state such that they are more receptive to excitatory inputs from the cortex.64 If this is so, information regarding emotionally significant cues is transmitted from the BLNG to the ventral striatum, facilitating incoming cortical inputs to this region (see Figure 4). Since pathways from auditory, visual, and somatosensory association cortices terminate in the BLNG,65 aberrant firing in this region would result in the transfer of such information, at inappropriate times, back to the cortex, or to the ventral striatum, or both.
Pathways and Connections
The organization of basal ganglia circuitry provides clues as to how information that is “tagged” with emotional content by the amygdala, and then transmitted to the ventral striatum (either directly or via amygdalo-corticostriatal paths), is translated into action. Understanding these connections provides a useful model for how, in psychotic states, limbic information that is inappropriately processed may give rise to bizarre behaviors. In primates, information flows from cortical regions (including the cortical-like BLNG of the amygdala) to the striatum and is relayed via the globus pallidus or the substantia nigra to the thalamus, and then back to cortex to close the loop. In general, these basal ganglia circuits are arranged topographically, in parallel, segregated loops.66,67 Thus, the striatum is divided into functional domains based on inputs from discrete cortical regions. The ventral striatum is considered limbic-related, receiving inputs from the BLNG of the amygdala, hippocampus, and anterior cingulate, orbitofrontal, and insular cortex.
This segregation of basal ganglia circuits changes at the level of the dopamine cells of the substantia nigra.68 Neuronal tracing studies in primates show that ventral striatonigral (limbic-related) inputs terminate across a wide extent of the dopamine cells of the substantia nigra pars compacta.69,70 This arrangement places the ventral striatal neurons, which receive amygdaloid inputs from the BLNG and other limbic regions, in a position to influence a large population of dopamine cells that project back to widespread functional domains of striatum, including dorsolateral (sensorimotor-related) striatum.71–73 In this way, limbic information from the BLNG and other limbic regions is channeled to the ventral striatum, and from there to a broad expanse of dopamine cells in the substantia nigra pars compacta. The dopamine neurons project back to modulate not only the ventral (limbic-related) striatum, but also the dorsolateral (sensorimotor-related) regions. Thus, the ventral striatum can modulate sensorimotor circuits via striatonigrostriatal loops. The reverse situation does not hold, in that dorsolateral (sensorimotor) striatonigral efferents terminate in a relatively small part of the nondopaminergic pars reticulata.69 This organizational schema may explain how the emotional and perceptual distortions mediated through limbic channels can influence motor output, with resulting bizarre actions.
Disrupted circuitry of the BLNG in the case of Ms. A. may be due in part to the hamartias detected in this brain region. The vulnerability of the BLNG—and other medial temporal lobe structures—to developmental anomalies is not well understood. Embryological studies suggest that, in humans, the BLNG cells migrate and differentiate later than cells in other divisions of the amygdala.74–76 The cells of the amygdala are derived from the periventricular germinal layer, and they first appear as early as the 6th week of life. However, the cells of the BLNG continue to develop into the second trimester, with the lateral nucleus of the BLNG undergoing intensive changes in histogenesis between 12 and 24 weeks.76 It is during this period of the second trimester that cells of the BLNG, the largest nuclear group of the amygdala, may be most vulnerable.
CONCLUSION
Schizophrenia and schizophrenia-like illnesses likely comprise a heterogeneous group of disorders with similar phenotypes. Thus, when we consider pathogenetic mechanisms, it is entirely possible that across patients different parts of the frontotemporal neural network are affected, possibly at critical periods in development. For example, abnormalities in the cytoarchitecture of prefrontal cortical regions may underlie chronic psychotic illness in some cases, whereas primary lesions in temporal lobe structures may underlie other forms of the illness. Furthermore, in some cases subtle brain dysgenesis may be a necessary but not sufficient condition for the expression of a schizophrenic phenotype. In such cases small “acquired” focal brain lesions may be critical events that compromise congenitally disrupted circuitry. The case of Ms. A. is informative precisely because the clinical data do not neatly fit either the category of schizophrenia or temporal lobe epilepsy. In this case, a neurobiologic perspective, rather than traditional clinical categories, may provide a better framework for understanding both the content and form of her symptoms. Neuropathologic evidence points to abnormalities in and near the left basolateral amygdala. We propose that in the case of Ms. A., abnormal firing of the basolateral amygdala fits as a pathogenic mechanism of her chronic psychosis, given the amygdala's extreme vulnerability to kindling, the timing of its embryologic development, and our current knowledge of the functional anatomy and physiology of this brain region.
ACKNOWLEDGMENTS
The authors acknowledge and thank Ms. Barbara Swigert for her support in the preparation of this manuscript. Dr. Fudge's work is supported in part by a National Research Service Award (MH18911) and by the Theodore and Vada Stanley Research Foundation.

FIGURE 1. Periventricular temporal white matter subjacent to area with hamartias and spheroid. Corpora amylacea are seen throughout as dense black granules (arrows). Luxol fast blue PAS, original ×40, reproduced at 82%.
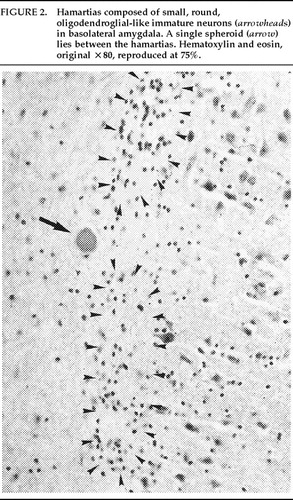
FIGURE 2. Hamartias composed of small, round, oligodendroglial-like immature neurons (arrowheads) in basolateral amygdala. A single spheroid (arrow) lies between the hamartias. Hematoxylin and eosin, original ×80, reproduced at 75%.
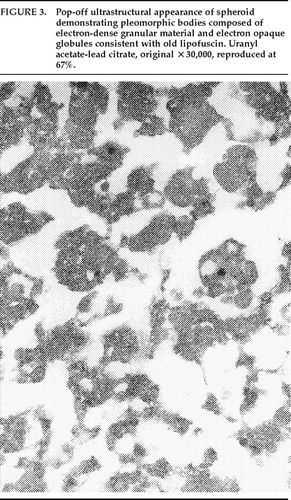
FIGURE 3. Pop-off ultrastructural appearance of spheroid demonstrating pleomorphic bodies composed of electron-dense granular material and electron opaque globules consistent with old lipofuscin. Uranyl acetate-lead citrate, original ×30,000, reproduced at 67%.
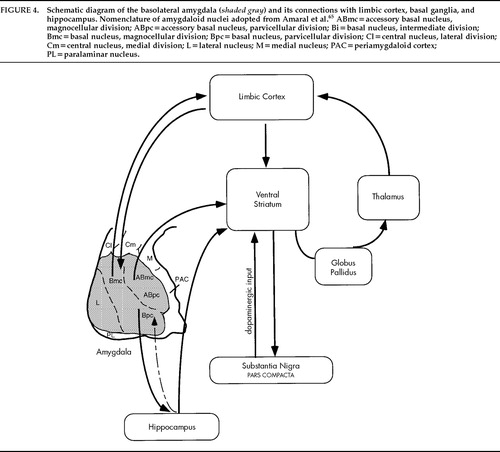
FIGURE 4. Schematic diagram of the basolateral amygdala (shaded gray) and its connections with limbic cortex, basal ganglia, and hippocampus. Nomenclature of amygdaloid nuclei adopted from Amaral et al.65 ABmc=accessory basal nucleus, magnocellular division; ABpc=accessory basal nucleus, parvicellular division; Bi=basal nucleus, intermediate division; Bmc=basal nucleus, magnocellular division; Bpc=basal nucleus, parvicellular division; Cl=central nucleus, lateral division; Cm=central nucleus, medial division; L=lateral nucleus; M=medial nucleus; PAC=periamygdaloid cortex; PL=paralaminar nucleus.
1. Torrey EF, Peterson MR: Schizophrenia and the limbic system. Lancet 1974; 2:942–946Crossref, Medline, Google Scholar
2. Stevens JR: An anatomy of schizophrenia? Arch Gen Psychiatry 1973; 29:177–189Google Scholar
3. Suddath RL, Casanova MF, Goldberg TE, et al: Temporal lobe pathology in schizophrenia: a quantitative magnetic resonance imaging study. Am J Psychiatry 1989; 146:464–472Crossref, Medline, Google Scholar
4. Bogerts B, Ashtari M, Degreef G, et al: Reduced temporal limbic structure volumes on magnetic resonance images in first episode schizophrenia. Psychiatry Res 1990; 35:1–13Crossref, Medline, Google Scholar
5. Weinberger DR: Schizophrenia and the frontal lobe. Trends Neurosci 1988; 11:367–370Crossref, Medline, Google Scholar
6. Goldman-Rakic PS: Working memory dysfunction in schizophrenia. J Neuropsychiatry Clin Neurosci 1994; 6:348–357Link, Google Scholar
7. Bilder RM, Bogerts B, Ashtari M, et al: Anterior hippocampal volume reductions predict frontal lobe dysfunction in first episode schizophrenia. Schizophr Res 1995; 17:47–58Crossref, Medline, Google Scholar
8. Weinberger DR, Berman KF, Suddath R, et al: Evidence of dysfunction of a prefrontal-limbic network in schizophrenia: a magnetic resonance imaging and regional cerebral blood flow study of discordant monozygotic twins. Am J Psychiatry 1992; 149:890–897Crossref, Medline, Google Scholar
9. Adams RD, Victor M, Mancall EL: Central pontine myelinolysis. Arch Neurol 1959; 81:154–172Google Scholar
10. Yachnis AT, Powell SZ, Olmsted JJ, et al: Distinct neurodevelopmental patterns of Bcl-2 and Bcl-x expression are altered in glioneuronal hamartias of the human temporal lobe. J Neuropathol Exp Neurol 1997; 56:186–198Crossref, Medline, Google Scholar
11. Strich SJ: Shearing of nerve fibers as a cause of brain damage due to head injury. Lancet 1961; 2:443–448Crossref, Google Scholar
12. Di Sant'Agnese PA, De Mesy-Jensen KL: Diagnostic electron microscopy on reembedded (“popped off”) areas of large Spurr epoxy sections. Ultrastruct Pathol 1984; 6:247–253Crossref, Medline, Google Scholar
13. Geschwind N: Disconnexion syndromes in animals and man. Brain 1965; 88:237–294Crossref, Medline, Google Scholar
14. Aggleton JP: The contribution of the amygdala to normal and abnormal emotional states. Trends Neurosci 1993; 16:328–333Crossref, Medline, Google Scholar
15. Morris JS, Frith CD, Perrett DI, et al: A differential neural response in the human amygdala to fearful and happy facial expressions. Nature 1996; 383(6603):812–815Google Scholar
16. Blanchard DC, Blanchard RJ: Innate and conditioned reaction to threat in rats with amygdaloid lesions. J Comp Physiol Psychol 1972; 81:281–290Crossref, Medline, Google Scholar
17. Nishijo H, Ono T, Nishino H: Single neuron responses in amygdala of alert monkey during complex sensory stimulation with affective significance. J Neurosci 1988; 8:3570–3583Crossref, Medline, Google Scholar
18. Klüver H, Bucy PC: Preliminary analysis of functions of the temporal lobes in monkeys. Arch Neurol Psychiatry 1939; 42:979–997Crossref, Google Scholar
19. Rosvold HE, Mirsky AF, Pribram KH: Influence of amygdalectomy on social behavior in monkeys. J Comp Physiol Psychol 1954; 47:173–178Crossref, Medline, Google Scholar
20. Weiskrantz L: Behavioral changes associated with ablation of the amygdaloid complex in monkeys. J Comp Physiol Psychol 1956; 49:381–391Crossref, Medline, Google Scholar
21. Bachevalier J: Medial temporal lobe structures and autism: a review of clinical and experimental findings. Neuropsychologia 1994; 32:627–648Crossref, Medline, Google Scholar
22. Aggleton JP: The functional effects of amygdala lesions in humans: a comparison with findings from monkeys, in The Amygdala: Neurobiological Aspects of Emotion, Memory, and Mental Dysfunction, edited by Aggleton JP. New York, Wiley-Liss, 1992, pp 485–503Google Scholar
23. Adolphs R, Tranel D, Damasio H, et al: Impaired recognition of emotion in facial expressions following bilateral damage to the human amygdala. Nature 1994; 372:669–672Crossref, Medline, Google Scholar
24. Halgren E, Walter RD, Cherlow DG, et al: Mental phenomena evoked by electrical stimulation of the human hippocampal formation and amygdala. Brain 1978; 101:83–117Crossref, Medline, Google Scholar
25. Heath RG, Monroe RR, Mickle WA: Stimulation of the amygdaloid nucleus in a schizophrenic patient. Am J Psychol 1954; 111:862–863Crossref, Google Scholar
26. Gloor P, Oliver A, Quesney LF, et al: The role of the limbic system in experimental phenomena of temporal lobe epilepsy. Ann Neurol 1982; 12:129–144Crossref, Medline, Google Scholar
27. Gloor P: Role of the amygdala in temporal lobe epilepsy, in The Amygdala: Neurobiological Aspects of Emotion, Memory, and Mental Dysfunction, edited by Aggleton JP. New York, Wiley-Liss, 1992, pp 505–538Google Scholar
28. Hill D: Psychiatric Disorders of Epilepsy. The Medical Press 1953; 229:473–475Google Scholar
29. Pond DA: Psychiatric aspects of epilepsy in children. Journal of the Indian Medical Profession 1957; 3:1441–1451Google Scholar
30. Slater E, Beard AW: The schizophrenia-like psychoses of epilepsy. Br J Psychiatry 1963; 109:95–150Crossref, Medline, Google Scholar
31. Flor-Henry P: Schizophrenic-like reactions and affective psychoses associated with temporal lobe epilepsy: etiological factors. Am J Psychiatry 1969; 126:400–404Crossref, Medline, Google Scholar
32. Stevens JR: Interictal clinical manifestations of complex partial seizures. Adv Neurol 1975; 11:85–112Medline, Google Scholar
33. Mendez MF, Grau R, Doss RC, et al: Schizophrenia in epilepsy: seizure and psychosis variables. Neurology 1993; 43:1073–1077Crossref, Medline, Google Scholar
34. Roberts GW, Done DJ, Bruton C, et al: A “mock up” of schizophrenia: temporal lobe epilepsy and schizophrenia-like psychosis. Biol Psychiatry 1990; 28:127–143Crossref, Medline, Google Scholar
35. Bruton CJ, Stevens JR, Frith CD: Epilepsy, psychosis, and schizophrenia: clinical and neuropathologic correlations. Neurology 1994; 44:34–42Crossref, Medline, Google Scholar
36. Sato M, Racine RJ, McIntyre DC: Kindling: basic mechanisms and clinical validity. Electroencephalogr Clin Neurophysiol 1990; 76:459–472Crossref, Medline, Google Scholar
37. Cain DP: Kindling and the amygdala, in The Amygdala: Neurobiological Aspects of Emotion, Memory, and Mental Dysfunction, edited by Aggleton JP. New York, Wiley-Liss, 1992, pp 539–560Google Scholar
38. Wolf HK, Campos MG, Zentner J, et al: Surgical pathology of temporal lobe epilepsy: experience with 216 cases. J Neuropathol Exp Neurol 1993; 52:499–506Crossref, Medline, Google Scholar
39. Falconer MA: Reversibility by temporal-lobe resection of the behavioral abnormalities of temporal-lobe epilepsy. N Engl J Med 1973; 289:451–455Crossref, Medline, Google Scholar
40. Savard G, Andermann F, Olivier A, et al: Postictal psychosis after partial complex seizures: a multiple case study. Epilepsia 1991; 32:225–231Crossref, Medline, Google Scholar
41. Taylor DC: Factors influencing the occurrence of schizophrenia-like psychosis in patients with temporal lobe epilepsy. Psychol Med 1975; 5:249–254Crossref, Medline, Google Scholar
42. Davies AM: The bcl-2 family of proteins, and the regulation of neuronal survival. Trends Neurosci 1995; 18:355–358Crossref, Medline, Google Scholar
43. Bruton CJ, Crow TJ, Frith CD, et al: Schizophrenia and the brain: a prospective clinico-neuropathological study. Psychol Med 1990; 20:285–304Crossref, Medline, Google Scholar
44. Stevens JR: Neuropathology of schizophrenia. Arch Gen Psychiatry 1982; 39:1131–1139Crossref, Medline, Google Scholar
45. Brown R, Colter N, Corsellis J, et al: Postmortem evidence of structural brain changes in schizophrenia. Arch Gen Psychiatry 1986; 43:36–42Crossref, Medline, Google Scholar
46. Crow TJ, Ball J, Bloom SR, et al: Schizophrenia as an anomaly of development of cerebral asymmetry. Arch Gen Psychiatry 1989; 46:1145–1150Crossref, Medline, Google Scholar
47. Roberts GW, Bruton CJ: Notes from the graveyard: neuropathology and schizophrenia. Neuropathol Appl Neurobiol 1990; 16:3–16Crossref, Medline, Google Scholar
48. Weinberger DR: From neuropathology to neurodevelopment. Lancet 1995; 346:552–557Crossref, Medline, Google Scholar
49. Pakkenberg B: Pronounced reduction of total neuron number in mediodorsal thalamic nucleus and nucleus accumbens in schizophrenics. Arch Gen Psychiatry 1990; 47:1023–1028Crossref, Medline, Google Scholar
50. Earle KM, Baldwin M, Penfield W: Incisural sclerosis and temporal lobe seizures produced by hippocampal herniation at birth. Arch Neurol Psychiatry 1953; 69:27–42Crossref, Google Scholar
51. Alheid GF, Heimer L: New perspectives in basal forebrain organization of special relevance for neuropsychiatric disorders: the striatopallidal, amygdaloid, and corticopetal components of substantia innominata. Neuroscience 1988; 27:1–39Crossref, Medline, Google Scholar
52. McDonald AJ, Beitz AJ, Larson AA, et al: Co-localization of glutamate and tubulin in putative excitatory neurons of the hippocampus and amygdala: an immunohistochemical study using monoclonal antibodies. Neuroscience 1989; 30:405–421Crossref, Medline, Google Scholar
53. McDonald AJ: Glutamate and aspartate immunoreactive neurons of the rat basolateral amygdala: colocalization of excitatory amino acids and projections to the limbic circuit. J Comp Neurol 1996; 365:367–379Crossref, Medline, Google Scholar
54. Amaral DG, Price JL: Amygdalo-cortical projections in the monkey (Macaca fascicularis). J Comp Neurol 1984; 230:465–496Crossref, Medline, Google Scholar
55. Llamas A, Avendano C, Reinoso-Suarez F: Amygdaloid projections to prefrontal and motor cortex. Science 1977; 195:794–796Crossref, Medline, Google Scholar
56. Russchen FT, Bakst I, Amaral DG, et al: The amygdalostriatal projections in the monkey: an anterograde tracing study. Brain Res 1985; 329:241–257Crossref, Medline, Google Scholar
57. Carmichael ST, Price JL: Limbic connections of the orbital and medial prefrontal cortex in macaque monkeys. J Comp Neurol 1996; 363:615–641Crossref, Google Scholar
58. Haber SN, Kunishio K, Mizobuchi M, et al: The orbital and medial prefrontal circuit through the primate basal ganglia. J Neurosci 1995; 15:4851–4867Crossref, Medline, Google Scholar
59. Kunishio K, Haber SN: Primate cingulostriatal projection: limbic striatal versus sensorimotor striatal input. J Comp Neurol 1994; 350:337–356Crossref, Medline, Google Scholar
60. Heimer L, Alheid GF, de Olmos JS, et al: The accumbens: beyond the core–shell dichotomy. J Neuropsychiatry Clin Neurosci 1997; 9:354–381Link, Google Scholar
61. Baldessarini RJ: Chemotherapy in Psychiatry: Principles and Practice, 2nd edition. Cambridge, MA, Harvard University Press, 1985Google Scholar
62. Deutch AY, Lee MC, Iadarola M: Regionally specific effects of atypical antipsychotic drugs on striatal fos expression: the nucleus accumbens shell as a locus of antipsychotic action. Molecular Cell Neuroscience 1992; 3:332–341Crossref, Medline, Google Scholar
63. Robertson GS, Fibiger HC: Neuroleptics increase c-fos expression in the forebrain: contrasting effects of haloperidol and clozapine. Neuroscience 1992; 46:315–328Crossref, Medline, Google Scholar
64. Moore H, Grace AA: Interactions between amygdala and prefrontal cortical afferents to the nucleus accumbens and their modulation by dopamine receptor activation (abstract). Society for Neuroscience Abstracts 1996; 22:1088Google Scholar
65. Amaral DG, Price JL, Pitkanen A, et al: Anatomical organization of the primate amygdaloid complex, in The Amygdala: Neurobiological Aspects of Emotion, Memory, and Mental Dysfunction, edited by Aggleton JP. New York, Wiley-Liss, 1992, pp 1–66Google Scholar
66. Alexander GE, Delong MR, Strick PL: Parallel organization of functionally segregated circuits linking basal ganglia and cortex. Annu Rev Neurosci 1986; 9:357–381Crossref, Medline, Google Scholar
67. Alexander GE, Crutcher MD: Functional architecture of basal ganglia circuits: neural substrates of parallel processing. Trends Neurosci 1990; 13:266–271Crossref, Medline, Google Scholar
68. Haber SN, Fudge JL: The primate substantia nigra and VTA: integrative circuitry and function. Crit Rev Neurobiol 1997; 11:323–342Crossref, Medline, Google Scholar
69. Lynd-Balta E, Haber SN: Primate striatonigral projections: a comparison of the sensorimotor-related striatum and the ventral striatum. J Comp Neurol 1994; 343:1–17Crossref, Medline, Google Scholar
70. Hedreen JC, Delong MR: Organization of striatopallidal, striatonigral, and nigrostriatal projections in the macaque. J Comp Neurol 1991; 304:569–595Crossref, Medline, Google Scholar
71. Somogyi P, Bolam JP, Totterdell S, et al: Monosynaptic input from the nucleus accumbens-ventral striatum region to retrogradely labelled nigrostriatal neurones. Brain Res 1981; 217:245–263Crossref, Medline, Google Scholar
72. Nauta WJH, Smith GP, Faull RLM, et al: Efferent connections and nigral afferents of the nucleus accumbens septi in the rat. Neuroscience 1978; 3:385–401Crossref, Medline, Google Scholar
73. Haber SN, Lynd-Balta E, Spooren WPTM: Integrative aspects of basal ganglia circuitry, in The Basal Ganglia, vol 4, edited by Percheron G, McKenzie JS, Féger J. New York, Plenum, 1994, pp 71–80Google Scholar
74. Humphrey T: The development of the human amygdala during early embryonic life. J Comp Neurol 1968; 132:135–166Crossref, Medline, Google Scholar
75. Muller F, O'Rahilly R: The human brain at stages 18–20, including the choroid plexuses and the amygdaloid and septal nuclei. Anat Embryol (Berl) 1990; 182:285–306Medline, Google Scholar
76. Nikolic I, Kostovic I: Development of the lateral amygdaloid nucleus in the human fetus: transient presence of discrete cytoarchitectonic units. Anat Embryol (Berl) 1986; 174:355–360Crossref, Medline, Google Scholar

