
Can Traumatic Brain Injury Cause Psychiatric Disorders?
Abstract
Traumatic brain injury (TBI) may cause psychiatric illness. This article reviews the evidence on the basis of an established set of causation criteria. The evidence is convincing for a strong association between TBI and mood and anxiety disorders. Substance abuse and schizophrenia are not strongly associated with TBI, and there is little research into the rates of personality disorders after TBI. Evidence for a biologic gradient is lacking, but such a gradient may not be relevant to TBI. Evidence for the correct temporal sequence is present. Preliminary evidence suggests a biologic rationale for TBI causing psychiatric illness. Further and methodologically improved research is supported and required.
Traumatic brain injury (TBI) has long been known to be associated with changes in mood, personality, and behavior.1–19 The existing research has also contributed to the hypothesis that factors related directly to the TBI may be causative of these changes. However, this research, for the most part, relied on dimensional rating of symptoms and did not include an assessment of the presence or absence of psychiatric disorders. The data generated by this research do, however, suggest that psychiatric disorders may be present at increased rates after TBI. TBI is considered by some to be a risk factor for psychiatric disorders.20,21
The establishment of a causative relationship between TBI and psychiatric disorders is important in terms of our understanding of these possible sequelae of TBI, and it will also help us in our understanding of the pathogenesis of these illnesses more generally. If it is shown that TBI causes psychiatric morbidity, this should alert clinicians to observe for, or to attempt to prevent, these outcomes. Such a finding of causation will also have a role in litigation related to outcomes after TBI; rather than finding, as is sometimes the case, that an individual's post-TBI difficulties are secondary to psychiatric disorder rather than being due to TBI, it would be appropriate to label the person's difficulties as being secondary to psychiatric disorder that in turn is secondary to TBI. Clearly establishing a causative role for TBI in producing psychiatric disorders is important from clinical, scientific, and legal perspectives.
Establishing an argument for causation of medical illness is often very difficult because the putative causative factor may be difficult to assess or may be confounded by the presence of other concurrent and potentially causative factors. This is certainly the case for TBI, in which the insult to the brain may be difficult to detect and may be accompanied by a host of other factors such as pain, losses, and hopelessness. It was not that long ago, however, that we also wondered about a causative relationship between newly discovered microscopic organisms grown in petri dishes and devastating epidemics of infectious diseases. This analogy is perhaps fitting, since clearly TBI may cause injury to the brain at the microscopic level and has been described as occurring at epidemic proportions.
How do we establish an argument for causation? Sir Bradford Hill22 proposed criteria for causation that include 1) consistently demonstrating an association between the causative agent and the purported outcome; 2) demonstrating a biologic gradient (i.e., that more of the causative agent causes more of the outcome); 3) demonstrating an appropriate temporal sequence (i.e., that the causative agent comes first in time); 4) providing a biologic rationale; 5) using analogous evidence (which is soft evidence in the sense that the pathophysiology of TBI is very different from that of any other neurological disorder); 6) finding experimental evidence (which, although the most compelling evidence for causation, will not be available for TBI because clearly it is unethical to experimentally induce a TBI in humans, and animal models of psychiatric illness are very limited); and 7) finding evidence of specificity of causation (this criterion has since been deemphasized, because even in infectious diseases, it is clear that a single organism may produce a number of diseases and some diseases may be produced by a number of infectious agents).
In this article we review the evidence available to support the hypothesis that TBI may cause some psychiatric disorders, using the most relevant of Hill's22 proposed criteria (criteria 1–4). Using these criteria is helpful because they 1) are widely accepted and applied throughout medicine, 2) increase rigor in establishing causation through the structure they provide, 3) facilitate teaching of important lessons about the role of the brain in producing psychiatric disorders (e.g., absence of a biologic gradient would suggest hypotheses related to brain function that might explain this finding), and 4) suggest research approaches (e.g., the need to establish a temporal sequence speaks to the need for prospective studies).
The Diagnostic and Statistical Manual of Mental Disorders23 was developed to provide a widely accepted, systematic, and reliable diagnostic scheme for psychiatric disorders. Currently the DSM-IV24 is used in psychiatry in North America, and the DSM or the International Classification of Diseases (ICD) is used elsewhere. Here we will emphasize research using the DSM or ICD diagnostic schema. The literature search was conducted in MEDLINE and included specific psychiatric disorders and “brain injury.” The review focuses on psychiatric disorders of most relevance to adults and on research done on adult populations. Focusing on disorders of childhood (e.g., attention-deficit/hyperactivity disorder) is of course highly relevant but is beyond the scope of this review. Including research involving exclusively children or adolescents is also inappropriate for this review because the developing brain may well differ in its response to injury in comparison to the adult brain. Similarly, disorders generally associated with elderly populations (e.g., dementia) also are not included in this review. MEDLINE searches of a number of neurotransmitters (associated with psychiatric illness) and TBI were also completed. To search for biologic mechanisms, existing reviews into the pathological changes accompanying TBI were reviewed.
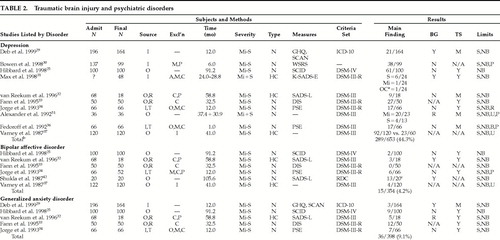
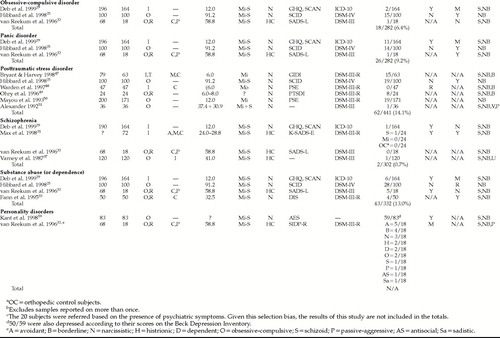
We initially summarize some of the main methodological limitations of the existing data. Each major psychiatric disorder for which there is some evidence is then reviewed in terms of the strength of the association, temporal sequence, and biologic gradient. This evidence is tabulated for each disorder. The coding system is summarized in Table 1 and the findings are presented in Table 2 and Table 2a. The prevalence data are then totaled (the limitations of this approach, given some of the methodological limitations discussed below, are acknowledged), and, as per the approach of Hibbard et al.,25 are contrasted with lifetime community prevalence data taken primarily from the Epidemiologic Catchment Area Survey.26 At the close of the discussion on each disorder, we review the evidence for biologic mechanisms through which TBI (or associated phenomena occurring at the time of, or as a result of, the TBI) may cause the specific disorder. For each disorder, we review the existing reviews as well as some original data from studies of these disorders in TBI. A section more generally discussing possible biologic mechanisms (derived from research into the neuronal and biochemical alterations caused by TBI) follows. We conclude with a brief discussion of future research needs.
METHODOLOGICAL LIMITATIONS OF THE EXISTING DATA
A quick glance at Table 2 and Table 2a will reveal many of the methodological limitations of the existing data. Many of the studies are naturalistic in nature and do not include a control group. Blindness cannot be ensured in this type of study, and researcher bias may affect data collection, analytical approach, and interpretation of the results. Selection biases may operate at many levels; for example, individuals who can be found, or who agree to the study, may be different from those who are not found or who refuse.
Many of the studies do not report the number of subjects with whom contact was attempted, nor the refusal rate. Their apparent admission N and their final N are hence, of necessity, reported as being equivalent in Table 2 and Table 2a. Corrigan et al.27 demonstrated that a psychiatric disorder may contribute to loss of subjects at follow-up; specifically, they found that a history of alcohol abuse and the alcohol blood level at the time of the TBI were both strongly associated with loss to follow-up status one year after TBI. It is conceivable that the presence of other psychiatric disorders may also affect recruitment rates for outcome studies after TBI.
Summarizing the data is made difficult by the multitude of recruitment strategies and sources. Diagnostic criteria for TBI, and for the severity of TBI, varied widely (data not tabulated). Some of the research is further compromised by unstructured outcome assessment or a brief follow-up period. Where structured assessments were used, the difficulty comes from the myriad of instruments selected. Even the DSM diagnoses may be invalid in TBI populations28 because TBI may conceivably mimic (as with concentration difficulties) or mask (e.g., frontal system damage producing expressive aprosody that may reduce the expression of sadness) psychiatric symptoms. Some of the research also did not assess for premorbid psychiatric status, thus limiting the assessment of temporal sequence, and some did not assess for a biologic gradient.
The validity of retrospective assessments of pre-TBI psychiatric histories has not been established. It is of course possible that either cognitive factors secondary to TBI or psychological factors related to the trauma of the event, or to the multiple ways that a TBI and associated injuries can affect an individual, may affect the recall of pre-TBI psychiatric histories.
The research into potential biologic mechanisms is limited by the number of variables assessed. It is always possible that the main etiologic factor was not well assessed or that an apparently causative agent is simply a marker for the true causative agent (i.e., that confounding may occur). Associations may also be missed because of a lack of power in some of the smaller studies. Finally, it should be noted that many of the findings related to biochemical alterations after TBI are derived from animal models of TBI, and as such may not accurately represent changes in humans.
STUDIES OF PSYCHIATRIC DISORDERS AND TRAUMATIC BRAIN INJURY
Major Depression
Ten studies assessing the prevalence of major depression (MD) following TBI were found.25,29–37 The study by Jorge et al.34 appears to have reported on a sample previously reported, and hence the data from this study are not included in the total. The study by Max et al.31 included only children and adolescents and is therefore also excluded from the total. It is included in the table to highlight to the reader that data informing us about MD in this population are available. (Additional important references are summarized by Max et al.31) MD was found to have occurred in 289 of 653 subjects (44.3%) over a period of less than 7.5 years following TBI. This contrasts with the general community population, in which the lifetime prevalence is 5.9%.26 Hence, TBI increases the risk of MD by a relative risk of at least 44.3/5.9=7.5. Clearly TBI significantly increases the risk of developing MD, and this result was fairly consistent across studies. The data regarding a biologic gradient are mixed, with some studies reporting evidence for such a gradient, others reporting that no gradient exists, and one suggesting that less severe TBI is associated with MD. The data related to the temporal sequence of MD in association with TBI were also mixed, with some studies strongly suggesting that MD follows TBI and others reporting that some of the sample had experienced MD prior to the TBI. None of the studies reported that MD consistently predated the TBI.
Alexander35 felt that MD may be “a reaction to failure at normal activities in the absence of any obvious neurological dysfunction” (p. 229) or that it may emerge out of postconcussion syndrome (PCS). Silver et al.21 review pre-DSM data that showed that depression was not related to severity of TBI but was associated with neuropsychological impairment and right hemisphere damage due to penetrating injuries. Robinson and Jorge38 review their research36 and conclude that premorbid psychiatric disorder and social impairments may contribute to MD following TBI. They also note that left dorsolateral frontal and left basal ganglia lesions are strongly associated with early MD and suggest that these sites may be important in eliciting biochemical responses that lead to depression. Finally, they note that MD is “not simply a psychological response to the severity of physical or intellectual impairment”38 (p. 237) but that impaired social functioning does seem to play a contributory role. Fann39 feels that the evidence supports a correlation between lesion location and emergence of psychiatric illness after TBI, but that the evidence does not support a biologic gradient. Rosenthal et al.28 note that noradrenergic and serotonergic projections from the brainstem enter the cortex by way of the frontal pole, and since this is a common site of contusion during TBI, they hypothesize that even “a small lesion in this area could potentially disrupt widespread cortical aminergic function”28 (p. 95). However, they feel that in general our knowledge about neurobiologic correlates of depression following TBI is limited and hence few conclusions can be drawn. Similarly, they conclude that there “are at present no experimental studies examining comprehensive psychosocial models and their proposed causal mechanisms of depression after TBI” (p. 95), while noting some of the retrospective data reviewed above by Robinson and Jorge.38 Finally, they note that the severity of depressive symptoms increases with the time since injury and with the degree of neuropsychological dysfunction.
Fann et al.33 found that the group of subjects with both depression and anxiety perceived themselves as being more ill, and functioning more poorly, than did the nondepressed anxious group. It is unclear whether depression with anxiety led to these altered perceptions of illness, or vice versa. The depressed and anxious group also had more symptoms of PCS. Deb et al.29 performed a logistic regression analysis with presence of any psychiatric disorder as the dependent variable; younger age, poorer TBI outcome (as measured by the Glasgow Outcome Scale), pre-TBI alcohol and psychiatric histories, lower Mini-Mental State Examination score, and lower number of years of education all entered the model. Jorge et al.34 found that the depressed group did not differ from the nondepressed group in terms of activities of daily living, cognitive functioning, or social functioning, nor did they differ in terms of their social supports. Logistic regression showed that depression was associated with left anterior CT scan lesions. Bowen et al.30 found that only pre-TBI occupational status was associated with MD; 60% of those not working before their TBI became depressed, versus 33% of those who were working. Van Reekum et al.32 found trend-level evidence of gender differences; 7 of 10 women, versus 2 of 8 men (P=0.06), became depressed post-TBI. Persinger40 analyzed MMPI data that suggested that “phasic or intermittent elevations of activity within limbic structures could be the primary etiology of depression”40 (p.[008]1286) post-TBI. Saran41 found that only 1 of 10 patients with melancholic depression after TBI had an abnormal dexamethasone suppression test, versus 91% of patients with primary melancholic depression, suggesting that melancholic depression post-TBI is not associated with hypothalamic-pituitary-adrenal axis dysfunction.
Bipolar Affective Disorder
Six studies have reported on 374 subjects after TBI;25,32–34,37,42 however, one of the studies42 was strongly affected by a selection bias (referrals to the study being dependent on the presence of psychiatric symptoms), so the data from this study (N=20) are not included. Bipolar affective disorder (BAD) occurred in 15 of the remaining 354 subjects (4.2%) over a maximum of 7.5 years of follow-up, and this contrasts with the general community lifetime prevalence rate of 0.8%.26 Hence the relative risk, as with MD, is large, at an estimated 4.2/0.8=5.3. Data regarding a biologic gradient is mixed, but the evidence for a temporal sequence, when assessed, was consistently positive.
Shukla et al.42 found that seizures were frequent, occurring in 50% of subjects, in their sample of 20 subjects who developed mania after closed TBI. Further evidence for a seizure hypothesis for secondary mania came from Pope et al.,43 who noted a preferential response to valproate, versus lithium, in BAD after TBI. Starkstein et al.44 found that 9 of 11 patients who developed manic syndromes after brain injury had right hemisphere involvement, and 8 of 11 had lesions involving the limbic system. Mean values for bifrontal and third-ventricle/brain ratios of the manic subjects were greater than those of nonmanic matched subjects. Five manic subjects had a family history of mood disorder. These data suggested that “the confluence of either anterior subcortical atrophy and a focal lesion of a limbic or limbic-connected region of the right hemisphere, or genetic loading and a limbic-connected right hemisphere lesion”44 (p. 1069) may account for mania after TBI. Jorge et al.34 found that mania after TBI was associated with temporal basal polar lesions and was not associated with severity of TBI, degree of physical or cognitive impairment, level of social functioning, or personal or family history of psychiatric disorder. van Reekum et al.32 found evidence of gender differences, with 1 of 10 females, versus 4 of 8 males (P=0.06), developing BAD or cyclothymia post-TBI.
Generalized Anxiety Disorder
Five studies25,29,32–34 have reported on the prevalence of generalized anxiety disorder (GAD) after TBI. In these studies, 36 of 398 subjects had GAD (9.1%) over a maximum of 7.5 years, leading to a relative risk of 9.1/4.025=2.3. Evidence for a temporal sequence was consistently positive, whereas that for a biologic gradient was mixed. Two studies reported evidence consistent with an absence of a biologic gradient; one was positive for a biologic gradient; and one suggested an inverse gradient exists.
Epstein and Ursano45 review the sparse literature related to anxiety disorders (in general) after TBI and conclude that “the interrelationships between anxiety and TBI are multifactorial, and the effect of specific tissue damage upon the nature of the symptomatology remains uncertain”45 (p. 306).
Obsessive-Compulsive Disorder
Three studies have reported on 282 subjects.25,29,32 Eighteen (6.4%) had obsessive-compulsive disorder (OCD) over a maximum of 7.5 years of follow-up, leading to an estimated relative risk of 6.4/2.526=2.6. Evidence for both a biologic gradient and temporal sequence was mixed.
Kant et al.46 reviewed the available literature, along with data they derived from 4 cases of OCD following mild TBI. While noting that the evidence showed inconsistencies, they concluded that the weight of the evidence nonetheless indicated a possible causative role for frontal system impairment.
Panic Disorder
Three studies25,29,32 have found rates of panic disorder averaging 9.2% (26/282) over a maximum of 7.5 years, yielding an estimated relative risk of 9.2/1.626=5.8. Evidence for a biologic gradient was mixed, but was more consistently positive for a temporal sequence. There are no available data to support pathophysiologic hypotheses in panic disorder after TBI.
Posttraumatic Stress Disorder
Six studies of 441 subjects25,47–51 revealed a rate of 62/441 (14.1%) over a maximum period of 7.5 years for posttraumatic stress disorder (PTSD), yielding an estimated relative risk of 14.1/8.052=1.8. Evidence for a biologic gradient was not provided in most studies; in those in which data are reported, the results were either negative or showed an inverse relationship between severity of TBI and rate of PTSD. Evidence for a temporal association was also rarely given. One study suggested that a temporal association does exist.
Mayou et al.50 found that PTSD is “not associated with a neurotic predisposition” but is “strongly associated with horrific memories of the accident”50 (p. 647). PTSD did not occur, in their sample, in subjects who lost consciousness during the TBI or who were amnestic for the event. Similarly, Warden et al.48 found that while 6 of 47 veterans (13%) who had suffered a moderate TBI and who were amnestic for the event developed avoidance and arousal criteria of PTSD, none developed the full syndrome, and none met the criteria of reexperiencing the event. Sbordone and Liter53 found that although PTSD patients, after a motor vehicle accident (MVA), a fall, or a blunt trauma, could fully recall the event, PCS patients could not. Ohry et al.49 found that women were predisposed to develop PTSD: 6 of 10 women, versus 2 of 14 men, developed PTSD following TBI. King54 reported a single case of a patient who suffered 2.5 days of posttraumatic amnesia after an MVA. Only a single “island” of memory was preserved, and that for a period immediately after the MVA when the patient was on the ground after having been thrown from the vehicle. Reexperiencing of this island of memory was felt to possibly contribute to the development of PTSD in this patient. Bryant and Harvey47 found that PTSD occurred in 82% of mild-TBI patients who had experienced acute stress disorder earlier (<1 month postinjury), but in only 11% of those who did not suffer acute stress disorder.
Schizophrenia
Schizophrenia (SCZ) appeared to be relatively uncommon in the four studies reporting data.29,31,32,37 When we excluded the study of children and adolescents by Max et al.,31 the resulting rate was 0.7% (2/302) over a maximum of 4.9 years of follow-up. This yields a relative risk of 0.7/1.526=0.5. Clearly these low numbers limit an assessment of the biologic gradient; however, the data did suggest that one may exist, since the cases were restricted to those with severe TBI (for those cases in which severity was reported). The evidence for a temporal sequence was mixed. Wilcox and Nasrallah55 performed a case-control study of SCZ and found that “head trauma” before age 10 years was more common in hospitalized SCZ subjects (22/200) than in those with BAD (6/122, P=0.06) or depression (3/203, P=0.0001) or in surgical control subjects (1/134, P=0.0001).
Smeltzer et al.56 reviewed the evidence related to anatomical localization of brain injury and relationship to psychosis. They found the evidence to be sparse, severely flawed, and inconsistent. O'Callaghan et al.57 reported on a patient with early-onset SCZ (age 16 years) who had sustained a blow to the left frontal-parietal region at age 14. A problem with this study, at least from the standpoint of localization, was the finding of generalized atrophy on CT scan. Buckley et al.58 reported on 3 patients with a history of cerebral trauma (loss of consciousness greater than 4 hours) and SCZ, and compared them with 2 patients with schizoaffective disorder and cerebral trauma. MRI scanning showed evidence of left temporal lobe abnormalities in all of the SCZ patients and in neither of the schizoaffective patients.
Substance Abuse
Substance abuse (SA) or dependence was common, at 13.0% (43/332), in the four reporting studies25,29,32,33 over a maximum follow-up period of 7.5 years. However, this rate is lower than that reported for the general community, with data suggesting a lifetime prevalence of substance abuse of 16.7%.26 The data from Deb et al.29 may have skewed this estimate, as they are reporting on substance dependence rather than substance abuse. Deleting their data yields a rate of 37/168=22.0%, and a relative risk of 22.0/16.7=1.3. Evidence for a biologic gradient, and for a temporal sequence, was mixed. There are no available data to support pathophysiologic hypotheses in substance abuse disorder after TBI.
Personality Disorders
Only one study has reported on DSM personality disorders, with avoidant, borderline, and narcissistic personality disorders being the most common.32 The numbers, however, are very low. Evidence for a biologic gradient was mixed, and the temporal sequence was not assessed. A categorical approach to the diagnosis of apathy59 is also reported on; this personality syndrome appears to be common after TBI and is associated with a biologic gradient. However, apathy is not recognized in the DSM series. The temporal sequence was not reported on in the study of apathy. Of note was the finding that apathy was found concurrently with MD in 50 of 59 cases. A full discussion of neurobiological issues related to personality change is beyond the scope of this paper, but frontal system involvement is frequently implicated.60–62
BIOLOGICAL MECHANISMS
Although pathophysiologic considerations related to each of the psychiatric disorders are discussed above, much can also be inferred from consideration of the pathophysiologic changes observed after TBI in general. Hume et al.63 note that diffuse axonal injury post-TBI is usually observed in the corpus callosum and in the brainstem. Levin et al.64 found that 17 of 20 subjects admitted for mild to moderate TBI had lesions on MRI, primarily involving the frontal and temporal regions. Alavi65 reviewed PET data that showed whole brain glucose hypometabolism post-TBI that correlated with the Glasgow Coma Scale score. Frontal region hypometabolism was also reported by Alavi.
Silver et al.66 note that TBI may cause contusional injuries affecting brain regions involved in the mediation of mood, especially “along the temporal lobes and frontal cortex” (p. 13), as well as diffuse axonal injury—which, in addition to disrupting neuronal circuits directly, may also disrupt neurotransmitter systems such as norepinephrine, serotonin, dopamine, and acetylcholine. Preliminary data that support the possibility of changes to these neurotransmitter systems were reviewed. Hypoxia may lead to free radical and excitotoxic neurotransmitter release, which cause further neuronal damage to these systems. Silver and Yudofsky67 note that several studies have reported neurochemical changes post-TBI, with indications that norepinephrine, serotonin, dopamine, and acetylcholine “are dramatically affected by TBI”67 (p. 637). Cholinergic deficits were shown to be associated with behavioral changes in moderately fluid-concussed rats.68 More recently Tanaka et al.69 found acute (i.e., at 25 seconds) increases in acetylcholine in concussed mice, as well as decreased norepinephrine, in the absence of changes to dopamine and serotonin. Tang et al.70 demonstrated that increased dopamine after mild TBI is associated with memory deficits in mice. Cerebrospinal fluid levels of substance P and serotonin were lower, and the levels of lipid peroxidation products were higher, in patients who had suffered a recent TBI versus healthy subjects having minor surgical procedures.71 The CSF changes did not correlate with the Glasgow Coma Scale score. In another study, increased serotonin levels in the extracellular fluid were also observed 10 minutes after brain trauma in rats.72 Increases in γ-aminobutyric acid have also been found, particularly in the dentate gyrus of brain-injured rats.73
Some of the changes to neurotransmitter systems may occur weeks after the initial TBI. Ciallella et al.74 found that there were no changes in vesicular acetylcholine transporter protein or in M2 receptors at 1 day and 1 week post-TBI in rats. At 2 and 4 weeks, however, a 40%–50% increase in vesicular acetylcholine transporter protein and a 25%–30% decrease in M2 receptors were observed. These changes particularly involved the hippocampus. These changes may have occurred in response to chronically lowered acetylcholine neurotransmission, which has also recently been demonstrated after TBI in rats.75
DISCUSSION
There are now a number of studies examining the issues related to DSM- or ICD-based psychiatric disorders after TBI. Although these studies have methodological limitations as discussed above, there is a strong and growing body of evidence to support the hypothesis that TBI frequently causes some, but not all, psychiatric disorders in those who have suffered a TBI. There is compelling evidence of causation for major depression, bipolar affective disorder, and the anxiety disorders after TBI. The evidence for psychosis and substance abuse suggests that TBI imposes either no increased risk or a very minor increased risk of these disorders. In the case of schizophrenia, the available research suggests that TBI may actually be protective for the disorder. However, psychosis is both a rare event and one that may be hard to detect, since many of these persons may have been receiving care in the psychiatric system, may be hard to find for other reasons, or may refuse to participate. These difficulties, in combination with the relatively small sample size currently available, suggests that this may be an inaccurate finding. Selection biases may also have limited the evaluation of risk associated with developing substance abuse disorders. There is very little research into the personality disorders, but the sparse research that does exist suggests that some of the personality disorders may also occur at high rates after TBI.
These results strongly support the need for a thorough and reliable assessment of mood, anxiety, and personality disorders in all persons who have suffered a TBI. Psychiatric illness, even in the absence of TBI, can cause impairment and disability contributing to handicap. Given the impact on functioning, and on subjective well-being, that these diagnoses can cause, there is a need to further study the impact of treatment of psychiatric disorders in the TBI population. Although we did not examine treatment in this article, few publications exist that address, with rigorous methodology, treatment issues of these disorders after TBI.
In terms of the prevalence of psychiatric disorders, major depression was the most common, at approximately 44% across all available studies. Bipolar illness was much less frequent, at approximately 4%. The anxiety disorders were common, ranging from approximately 6.5% for OCD to a high of approximately 14% for PTSD. Substance abuse was also fairly common, at 22%, while psychosis was uncommon at less than 1%. Little research was available that contrasted these rates with a control population. Estimate of the relative risk (RR) of psychiatric disorders, based on comparisons with community rates, is potentially problematic. Use of these data, however, generated estimates of relative risk as follows. The highest relative risk was for major depression, with an RR of 7.5. Bipolar disorder also had a high RR of 5.3. The RRs for the anxiety disorders clustered around an approximate figure of 2.0, with the exception of panic disorder with an RR of 5.8. The RRs for schizophrenia and substance abuse were close to or less than 1.0, suggesting either no, or a minor, increased risk for these disorders.
As discussed previously, the most rigorous data demonstrating an association between TBI and psychiatric disorders will derive from properly controlled studies. We found only three controlled studies. The study by Max et al.31 involved only children and adolescents, and that of van Reekum.32 studied a control group with known psychiatric pathology (these data were derived from a larger study of borderline personality disorder). Hence, only the study by Varney et al.37 provided well-controlled data in adults. They found an RR of 2.0 for MD. RRs for schizophrenia and bipolar affective disorder could not be calculated because no cases were found in the back-injured control population. Hence, the data of Varney et al. are generally supportive of the naturalistic studies, but the data related to MD suggest that relative risk estimates generated by comparison with the community may inflate the relative risk versus that generated by comparison with other injured populations. Demonstration of an association between the disorders with high RR and TBI meets the main criteria for causation but does not in and of itself establish causation.
Sir Bradford Hill's22 remaining criteria are now discussed. The evidence for a biologic gradient, in which increased severity of the TBI is associated with increased risk of psychiatric disorders, was mixed for all disorders with the exception of PTSD, for which there was compelling evidence of an inverse gradient (i.e., increased risk of PTSD with milder TBI). This would seem to weaken the argument for causation based on this criterion. As discussed above, though, it is possible that even small or minor injury occurring in critical areas of the brain may disrupt widespread neuronal systems and/or have dramatic effects on neurotransmitter systems. Further, it may be that our approach to assessing the severity of TBI (at present based largely on measures of the initial severity of the TBI, such as depth and duration of coma) may be fundamentally limited. It may also be that more severe TBI is protective for some psychiatric disorders via other mechanisms, such as reduced insight or other direct effects on brain systems involved in the production of these disorders. Hence the absence of a biologic gradient does not lessen the argument for causation. Indeed, the apparent lack of a biologic gradient for psychiatric disorders post-TBI is in and of itself an important clinical finding. If true, this finding implies that all individuals, regardless of the initial severity of the TBI, may be at risk of developing many of the psychiatric disorders reviewed herein. As regards the special case of PTSD, the finding of an apparent inverse relationship, as well as some of the evidence reviewed previously, strongly suggests that recalling the event is crucial for development of the full disorder. However, there is also evidence that TBI, regardless of severity, may increase the risk of developing PTSD symptoms by contributing to the production of increased arousal and avoidance behaviors.
In terms of the temporal sequence, it is clear from the data that some psychiatric disorders were present in some patients prior to the TBI. However, it is also clear that many more subjects had apparent onset of their disorders after the TBI. Furthermore, there was no consistent demonstration that pre-TBI psychiatric illness was a strong risk factor for post-TBI psychiatric illness. For the most part the temporal sequence criterion is being satisfied, although some of the available data suggest that pre-TBI psychiatric status may also be a risk factor for some post-TBI psychiatric disorders. What is also clear from the research, however, is that some of the psychiatric illnesses may have their onset months to years after the TBI, and this may seem to lower the evidence for causation. Some of the neurotransmitter research reviewed above suggests at least one possible mechanism in this regard, since it is increasingly clear that biochemical changes to the brain may occur at some period of time after the TBI.
In terms of biologic plausibility, there has been considerable research related to the mood disorders after TBI, but much less so for the other psychiatric disorders. There certainly appears to be a growing body of evidence related to changes in the brain that are strongly associated with psychiatric illness, and the research is suggesting some tantalizing leads into the operative biologic mechanisms in psychiatric illness after TBI. Nonetheless, this is an area that requires a significant expansion of research. Although the data were not reviewed herein, there is a strong biologic argument that personality disorders can be caused by TBI, but this causation argument is limited by the small number of prevalence studies and the absence of data regarding temporal sequence or biologic gradient.
Further research into the hypothesis that TBI causes psychiatric illness is certainly strongly supported by the existing data and is much needed. Methodological limitations of the existing data need to be addressed in future research; however, it is likely that doing so will be very difficult. For example, addressing some of the selection biases and loss-to-follow-up issues is challenging. It is obvious that appropriate control samples are required, but which group is best and most feasible? Spinal cord injury controls may be ideal from many points of view; however, some members of this group may also have had a TBI during the traumatic event, and this would carefully need to be assessed. Furthermore, how can one ever be sure, based on retrospective assessments, of the pre-TBI psychiatric history and presence of pre-TBI psychiatric risk factors? And yet how does one feasibly perform a prospective study? A large study over many years of follow-up of a high-risk (for TBI) sample would be best, but may be unfeasible. Another approach would be to document the pre-TBI history immediately after the TBI to minimize the risk of recall biases developing as the individual begins to become more aware of the outcome of the TBI over time. The challenge here, of course, will be to perform this assessment in the period when cognitive impairments are likely to be at their most severe. Family members as informants will almost certainly be needed. Ensuring blindness to TBI status at the time of psychiatric outcome assessment will also be difficult. Assessing TBI severity as related to the biologic gradient criterion is problematic, especially because small or minor lesions may have dramatic effects on brain function. Ultimately, research investigating the function of the brain in association with psychiatric illness is more likely to be fruitful than research relying on crude measures of severity such as the depth and duration of coma. Finally, it is obvious that much more research is required into the biological and psychosocial contributors to psychiatric illness in these populations. A comprehensive approach examining multiple probable contributing factors is needed. The research should be guided by some of the clues reviewed above and by the research into the pathogenesis of primary psychiatric illness.
ACKNOWLEDGMENTS
Dr. van Reekum receives support from the Kunin-Lunenfeld Applied Research Unit of Baycrest Centre and is engaged in research supported by the Alzheimer's Society of Canada.
 |
 |
 |
1 Dikmen S, Reitan RM: Emotional sequelae of head injury. Ann Neurol 1977; 2:492–494Crossref, Medline, Google Scholar
2 Levin HS, Grossman RG: Behavioral sequelae of closed head injury. Arch Neurol 1978; 35:720–727Crossref, Medline, Google Scholar
3 Lewin W, Marshall TF, Roberts AH: Long-term outcome after severe head injury. British Medical Journal 1979; 2:1533–1538Google Scholar
4 Weddell R, Oddy M, Jenkins D: Social adjustment after rehabilitation: a two year follow-up of patients with severe head injury. Psychol Med 1980; 10:257–263Crossref, Medline, Google Scholar
5 McKinlay WW, Brooks DN, Bond MR, et al: The short-term outcome of severe blunt head injury as reported by the relatives of the injured person. J Neurol Neurosurg Psychiatry 1981; 44:527–533Crossref, Medline, Google Scholar
6 Barth JT, Macciocchi SN, Giordani B, et al: Neuropsychological sequelae of minor head injury. Neurosurgery 1983; 13:529–533Crossref, Medline, Google Scholar
7 Fordyce DJ, Roueche JR, Prigatano GP: Enhanced emotional reaction in chronic head trauma patients. J Neurol Neurosurg Psychiatry 1983; 46:620–624Crossref, Medline, Google Scholar
8 Oddy M, Coughlan T, Tyerman A, et al: Social adjustment after closed head injury: a further followup seven years after injury. J Neurol Neurosurg Psychiatry 1985; 48:564–568Crossref, Medline, Google Scholar
9 Klonoff PS, Snow WG, Costa LD: Quality of life in patients 2 to 4 years after closed head injury. Neurosurg 1986; 19:735–743Crossref, Medline, Google Scholar
10 Brooks N, Campsie L, Symington C, et al: The five year outcome of severe blunt head injury: a relative's view. J Neurol Neurosurg Psychiatry 1986; 49:764–770Crossref, Medline, Google Scholar
11 Diamond R, Barth JT, Zillmer BA: Emotional correlates of mild closed head trauma: the role of the MMPI. Int J Clin Neuropsychol 1988; 10:35–40Google Scholar
12 Schoenhuber R, Gentilini M: Anxiety and depression after mild head injury: a case control study. J Neurol Neurosurg Psychiatry 1988; 51: 722–724Google Scholar
13 Blackerby WF, Porter S: Psychosocial aspects of sexual dysfunction in head injury. Physical Medicine and Rehabilitation 1989; 3:143–155Google Scholar
14 Bornstein RA, Miller HB, VanSchoor JT: Neuropsychological deficit and emotional disturbance in head injured patients. J Neurosurg 1989; 70:509–513Crossref, Medline, Google Scholar
15 MacNiven E, Finlayson MAJ: The interplay between emotional and cognitive recovery after closed head injury. Brain Inj 1993; 7:241–246Crossref, Medline, Google Scholar
16 Sherman AG, Shaw TG, Glidden H: Emotional behavior as an agenda in neuropsychological evaluation. Neuropsychol Rev 1994; 4:45–69Crossref, Medline, Google Scholar
17 Parker RS: The spectrum of emotional distress and personality changes after minor head injury incurred in a motor vehicle accident. Brain Inj 1996; 10:287–302Crossref, Medline, Google Scholar
18 Lannoo E, De Deyne C, Colardyn F, et al: Personality change following head injury: assessment with the Neo Five-Factor Inventory. J Psychosom Res 1997; 43:505–511Crossref, Medline, Google Scholar
19 Kurtz JE, Putnam SH, Stone C: Stability of normal personality traits after traumatic brain injury. J Head Trauma Rehabil 1998; 13:1–14Crossref, Medline, Google Scholar
20 Gualtieri CT, Cox DR: The delayed neurobehavioral sequelae of traumatic brain injury. Brain Inj 1991; 5:219–232Crossref, Medline, Google Scholar
21 Silver JM, Hales RE, Yudofsky SC: Neuropsychiatric aspects of traumatic brain injury, in The American Psychiatric Press Textbook of Neuropsychiatry, 2nd edition, edited by Yudofsky SC, Hales RE. Washington, DC, American Psychiatric Press, 1992, pp 363–395Google Scholar
22 Hill AB: The environment and disease: association or causation? Proc R Soc Med 1965; 58:293–300Google Scholar
23 American Psychiatric Association: Diagnostic and Statistical Manual of Mental Disorders, 3rd edition. Washington, DC, American Psychiatric Association, 1980Google Scholar
24 American Psychiatric Association: Diagnostic and Statistical Manual of Mental Disorders, 4th edition. Washington, DC, American Psychiatric Association, 1994Google Scholar
25 Hibbard MR, Uysal S, Kepler K, et al: Axis 1 psychopathology in individuals with traumatic brain injury. J Head Trauma Rehabil 1998; 13:24–39Crossref, Medline, Google Scholar
26 Bourdon KH, Rae DS, Locke BZ, et al: Estimating the prevalence of mental disorders in U.S. adults from the Epidemiologic Catchment Area Survey. Public Health Rep 1992; 107:663–668Medline, Google Scholar
27 Corrigan JD, Bogner JA, Mysiw WJ, et al: Systematic bias in outcome studies of persons with traumatic brain injury. Arch Phys Med Rehabil 1997; 78:132–137Crossref, Medline, Google Scholar
28 Rosenthal M, Christensen BK, Ross TP: Depression following traumatic brain injury. Arch Phys Med Rehabil 1998; 79:90–103Crossref, Medline, Google Scholar
29 Deb S, Lyons I, Koutzoukis C, et al: Rate of psychiatric illness 1 year after traumatic brain injury. Am J Psychiatry 1999; 156:374–378Medline, Google Scholar
30 Bowen A, Neumann V, Conner M, et al: Mood disorders following traumatic brain injury: identifying the extent of the problem and the people at risk. Brain Inj 1998; 12:177–190Crossref, Medline, Google Scholar
31 Max JE, Koele SL, Smith Jr WL, et al: Psychiatric disorders in children and adolescents after severe traumatic brain injury: a controlled study. J Am Acad Child Adolesc Psychiatry 1998; 37:832–840Crossref, Medline, Google Scholar
32 van Reekum R, Bolago I, Finlayson MA, et al: Psychiatric disorders after traumatic brain injury. Brain Inj 1996; 10:319–327Crossref, Medline, Google Scholar
33 Fann JR, Katon WJ, Uomoto JM, et al: Psychiatric disorders and functional disability in outpatients with traumatic brain injury. Am J Psychiatry 1995; 152:1493–1499Google Scholar
34 Jorge RE, Robinson RG, Starkstein SE, et al: Depression and anxiety following traumatic brain injury. J Neuropsychiatry Clin Neurosci 1993; 5:369–374Link, Google Scholar
35 Alexander MP: Traumatic brain injury, in Psychiatric Aspects of Neurological Disease, edited by Benson DF, Blumer D. New York, Grune and Stratton, 1975, pp 219–249Google Scholar
36 Fedoroff JP, Starkstein SE, Forrester AW, et al: Depression in patients with acute traumatic brain injury. Am J Psychiatry 1992; 149:918–923Crossref, Medline, Google Scholar
37 Varney NR, Martzke JS, Roberts RJ: Major depression in patients with closed head injury. Neuropsychology 1987; 1:7–9Crossref, Google Scholar
38 Robinson RG, Jorge R: Mood disorders, in Neuropsychiatry of Traumatic Brain Injury, edited by Silver JM, Yudofsky SC, Hales RE. Washington, DC, American Psychiatric Press, 1994, pp 219–250Google Scholar
39 Fann JR: Traumatic brain injury and psychiatry. J Psychosom Res 1997; 43:335–343Crossref, Medline, Google Scholar
40 Persinger MA: Depression following brain trauma is enhanced in patients with mild discrepancies between intelligence and impairment on neuropsychological scores. Percept Mot Skills 1997; 84:1284–1286Google Scholar
41 Saran AS: Depression after minor closed head injury: role of dexamethasone suppression test and antidepressants. J Clin Psychiatry 1985; 46:335–338Medline, Google Scholar
42 Shukla S, Cook B, Mukherjee S, et al: Mania following head trauma. Am J Psychiatry 1987; 144:93–96Crossref, Medline, Google Scholar
43 Pope Jr. HG, McElroy SL, Satlin A, et al: Head injury, bipolar disorder and response to valproate. Compr Psychiatry 1988; 29:34–38Crossref, Medline, Google Scholar
44 Starkstein SE, Pearlson GD, Boston J, et al: Mania after brain injury: a controlled study of causative factors. Arch Neurol 1987; 44:1069–1073Google Scholar
45 Epstein RS, Ursano RJ: Anxiety disorders, in Neuropsychiatry of Traumatic Brain Injury, edited by Silver JM, Yudofsky SC, Hales RE. Washington, DC, American Psychiatric Press, 1994, pp 285–311Google Scholar
46 Kant R, Smith-Seemiller L, Duffy JD: Obsessive Compulsive disorder after closed head injury: review of literature and report of four cases. Brain Inj 1996; 10:55–63Crossref, Medline, Google Scholar
47 Bryant RA, Harvey AG: Relationship between acute stress disorder and posttraumatic stress disorder following mild traumatic brain injury. Am J Psychiatry 1998; 155:625–629Crossref, Medline, Google Scholar
48 Warden DL, Labbate LA, Salazar AM, et al: Posttraumatic stress disorder in patients with traumatic brain injury and amnesia for the event? J Neuropsychiatry Clin Neurosci 1997; 9:18–22Google Scholar
49 Ohry A, Rattok J, Soloman Z: Post-traumatic stress disorder in brain injury patients. Brain Inj 1996; 10:687–695Crossref, Medline, Google Scholar
50 Mayou R, Bryant B, Duthie R: Psychiatric consequences of road traffic accidents. BMJ 1993; 307:647–651Crossref, Medline, Google Scholar
51 Alexander MP: Neuropsychiatric correlates of persistent postconcussive syndrome. J Head Trauma Rehabil 1992; 7:60–69Crossref, Google Scholar
52 Kessler RC, Sonnega A, Bromet E et al: Posttraumatic stress disorder in the National Comorbidity Survey. Arch Gen Psychiatry 1995; 52:1048–1060Google Scholar
53 Sbordone RJ, Liter JC: Mild traumatic brain injury does not produce post-traumatic stress disorder. Brain Inj 1995; 9:405–412Crossref, Medline, Google Scholar
54 King NS: Post-traumatic stress disorder and head injury as a dual diagnosis: ”islands” of memory as a mechanism. J Neurol Neurosurg Psychiatry 1997; 62:82–84Crossref, Medline, Google Scholar
55 Wilcox JA, Nasrallah HA: Childhood head trauma and psychosis. Psychiatry Res 1987; 21:303–307Crossref, Medline, Google Scholar
56 Smeltzer DJ, Nasrallah HA, Miller SC: Psychotic disorders, in Neuropsychiatry of Traumatic Brain Injury, edited by Silver JM, Yudofsky SC, Hales RE. Washington, DC, American Psychiatric Press, 1994, pp 251–283Google Scholar
57 O'Callaghan E, Larkin C, Redmond O, et al: Early onset schizophrenia after teenage head injury: a case report with magnetic resonance imaging. Br J Psychiatry 1988; 153:394–396Crossref, Medline, Google Scholar
58 Buckley P, Stack JP, Madigan C, et al: Magnetic resonance imaging of schizophrenia-like psychoses associated with cerebral trauma: clinicopathological correlates. Am J Psychiatry 1993; 150:146–148Crossref, Medline, Google Scholar
59 Kant R, Duffy JD, Pivovarnik A: Prevalence of apathy following head injury. Brain Inj 1998; 12:87–92Crossref, Medline, Google Scholar
60 Starkstein SE, Robinson RG: Mechanism of disinhibition after brain lesions. J Nerv Ment Dis 1997; 185:108–114Crossref, Medline, Google Scholar
61 van Reekum R: Acquired and developmental brain dysfunction in borderline personality disorder. Can J Psychiatry 1993; 38(suppl l):S4–S10Google Scholar
62 Stuss DT, van Reekum R, Murphy KJ: Apathy states, in Neuropsychology of Emotion, edited by Borod J. New York, Oxford University Press (in press)Google Scholar
63 Hume Adams J, Doyle D, Graham DI, et al: Microscopic diffuse axonal injury in cases of head injury. Med Sci Law 1985; 25:265–269Crossref, Medline, Google Scholar
64 Levin HS, Amparo E, Eisenberg HM, et al: Magnetic resonance imaging and computerized tomography in relation to the neurobehavioral sequelae of mild and moderate head injuries. J Neurosurg 1987; 66:706–713Crossref, Medline, Google Scholar
65 Alavi A: Functional and anatomic studies of head injury. J Neuropsychiatry Clin Neurosci 1989; 1(suppl 1):S45–S50Google Scholar
66 Silver JM, Yudofsky SC, Hales RE: Depression in traumatic brain injury. Neuropsychiatry Neuropsychol Behav Neurol 1991; 4:12–23Google Scholar
67 Silver JM, Yudofsky SC: Psychopharmacology, in Neuropsychiatry of Traumatic Brain Injury, edited by Silver JM, Yudofsky SC, Hales RE. Washington, DC, American Psychiatric Press, 1994, pp 631–670Google Scholar
68 Robinson SE, Martin RM, Davis TR, et al: The effect of acetylcholine depletion on behavior following traumatic brain injury. Brain Res 1990; 509:41–46Crossref, Medline, Google Scholar
69 Tanaka K, Ogawa N, Asanuma M et al: Thyrotropin releasing hormone prevents abnormalities of cortical acetylcholine and monoamines in mice following head injury. Regul Pept 1997; 70:173–178Crossref, Medline, Google Scholar
70 Tang YP, Noda Y, Nabeshima T: Involvement of activation of dopaminergic neuronal system in learning and memory deficits associated with experimental mild traumatic brain injury. Eur J Neurosci 1997; 9:1720–1727Google Scholar
71 Karakucuk EI, Pasaoglu H, Pasaoglu A, et al: Endogenous neuropeptides in patients with acute traumatic head injury, II: changes in the levels of cerebrospinal fluid substance P, serotonin and lipid peroxidation products in patients with head trauma. Neuropeptides 1997; 31:259–263Crossref, Medline, Google Scholar
72 Busto R, Dietrich WD, Globus MY, et al: Extracellular release of serotonin following fluid-percussion brain injury in rats. J Neurotrauma 1997; 14:35–42Crossref, Medline, Google Scholar
73 Reeves TM, Lyeth BG, Phillips LL, et al: The effects of traumatic brain injury on inhibition in the hippocampus and dentate gyrus. Brain Res 1997; 757:119–132Crossref, Medline, Google Scholar
74 Ciallella JR, Yan HQ, Ma X, et al: Chronic effects of traumatic brain injury on hippocampal vesicular acetylcholine transporter and M2 muscarinic receptor protein in rats. Exp Neurol 1998; 152:11–19Crossref, Medline, Google Scholar
75 Dixon CE, Ma X, Marion DW: Reduced evoked release of acetylcholine in the rodent neocortex following traumatic brain injury. Brain Res 1997; 749:127–130Crossref, Medline, Google Scholar

