
Changes in Relative Glucose Metabolic Rate Following Cortisol Administration in Aging Veterans with Posttraumatic Stress Disorder: An FDG-PET Neuroimaging Study
The aim of our present study was to examine central effects of glucocorticoids on relative glucose metabolic rate (rGMR) in the hippocampus, dorsal amygdala, ventral amygdala, and anterior cingulate cortex (ACC). These regions have been implicated in PTSD pathophysiology 13 – 16 and are known to be regulated by glucocorticoids. 17 – 19 Studies examining activity of these regions of interest in response to presentation of traumatic or emotional stimuli have demonstrated alterations in metabolic activity of these structures as well as in their interconnections. 20 – 35 In particular, amygdala activation has most often been demonstrated to be increased in PTSD, while ACC activity is diminished. 20 – 24 , 36 However these differences vary by psychological task, with ACC activation in PTSD greater than in comparison subjects during trauma script tasks. 37 Because the ACC normally inhibits amygdala activity in response to stress, the neuroimaging findings support an impaired regulation of the neural circuitry connecting the amygdala to the ACC, possibly observed even under baseline conditions.
The amygdala projections leading to the ACC originate in the dorsal region of the amygdala, whereas the ventral amygdala, comprised of the basolateral complex, projects to orbitofrontal regions. 34 The basolateral complex has been specifically implicated in glucocorticoid enhancement of memory, 19 as shown on fMRI studies, 30 , 34 justifying the separate examination of the dorsal and ventral regions in PTSD.
In this study, brain metabolism was examined in response to placebo and an intravenous bolus of HCORT, administered in a randomized, double-blind manner, and quantified using [ 18 F]fluorodeoxyglucose (FDG) positron emission tomography (PET) neuroimaging. We hypothesized a greater response to the effects of HCORT in the PTSD+ group, consistent with enhanced responsiveness to glucocorticoids. In view of findings implicating laterality differences in PTSD, including reports of either greater group differences by hemisphere, or difference confined to one hemisphere, 20 , 22 – 24 we further hypothesized that placebo rGMR might differ according to hemisphere based on group, and to the extent that this would be true, the effects of HCORT might also differ by hemisphere.
METHODS
Participants
The sample consisted of 32 male veterans 52 to 81 years old (16 with chronic PTSD, 16 without PTSD) and represent a slightly larger sample than that for whom we previously reported an enhanced ACTH response 38 and improved performance on working memory following HCORT administration. 9 This lower age limit was imposed to compare the current findings with the only other report of the effects of HCORT on brain glucose metabolism, which was done in an aging population. 39 Furthermore, this age range is of interest because glucocorticoid-dependent memory deficits have been found to be particularly prominent in older persons with PTSD and appear to become more marked with age. 40 The study was approved by the institutional review boards at both the Mount Sinai School of Medicine and the U.S. Department of Veterans Affairs (VA). All participants provided written, informed consent prior to their participation.
Exclusion criteria were current or lifetime history of bipolar disorder, psychotic illness, organic mental disorder, current alcohol or drug abuse or dependence, neurological illness including cerebrovascular accident, head injury with loss of consciousness lasting greater than 10 minutes, or laboratory data indicating significant medical illness. Participants were abstinent from alcohol abuse for a minimum of 6 months and alcohol dependence for a minimum of 2 years, an adequate interval for recovery from the nonpermanent, neurotoxic effects of alcohol, 41 – 42 and were not taking psychiatric medications or steroids. The period of abstinence from alcohol abuse and dependence ranged from 2.5 to 43 years.
Clinical Assessments
Axis I diagnoses were made by trained psychologist raters according to the DSM-IV criteria using the Structured Clinical Interview for DSM-IV 43 and the Clinician Administered PTSD Scale (CAPS), 44 confirmed at a consensus conference.
PET Imaging After HCORT Versus Placebo Administration
Participants were instructed to report to the laboratory at 8 a.m. on two separate occasions, 1–2 weeks apart. A 20 gauge angiocath was placed in the antecubital region for blood sampling and delivery of either 17.5 mg HCORT or placebo (normal saline) and 18 FDG. A baseline blood sample was collected 10 minutes before the injection of saline or HCORT and at several intervals postinjection for ACTH and cortisol determination (data presented elsewhere 38 ). The dose of HCORT yielded circulating plasma cortisol levels within a high, but physiological range (mean ± standard error = 41.35±1.83 μg/dl). 38 To control for order effects, the occurrence of drug and placebo was randomly assigned in each of the two groups on the first session and was then counterbalanced in each group separately by alternating between placebo and cortisol day. Neither the rater nor the subject knew whether drug or placebo was injected.
FDG was administered intravenously 5 minutes after saline or HCORT. Positron emission tomography scans were carried out following a 35-minute uptake period, as described elsewhere, 45 on a General Electric Medical Systems scanner (model 2048) with in-plane resolution, 4.5 mm full width at half maximum (FWHM) 6.5 mm axially, in the Mount Sinai School of Medicine Neuroscience PET Lab. Twenty axial slices were obtained at 6.5 mm intervals. An individually molded thermoplastic face mask was used for each participant to help him remain stationary during image acquisition. Following the scan a series of neuropsychological tests were administered (results reported elsewhere 9 ).
MRI
MRI data were obtained on a 3T Siemens Allegra system. High-resolution structural images with good gray/white matter contrast were obtained using a T1-weighted Magnetization Prepared Rapid Gradient Echo sequence. A total of 208 slices were collected with isotropic resolution of 0.82 mm 3 , matrix size=256×256×208, field of view=21 cm, repetition time=2500 ms, echo time=4.38 ms, interval time=1100 ms and a 8° flip angle fast low angle shot acquisition giving a total imaging time of about 10 minutes. The magnetic resonance images had the skull removed with the Brain Extraction Tool and were then resectioned to standard Talairach coordinates using a rigid body 6 parameter transformation with FLIRT (Functional-Magnetic-Resonance-Imaging-Brain Linear Image Registration Tool v5). 46 Brain edges were visually traced on all MRI axial slices.
The procedure for tracing the hippocampus and amygdala are described in detail elsewhere. 47 – 48 Briefly, outlining of the amygdala begins at its largest extent (approximately the center in the anteroposterior dimension) where clear boundaries between gray matter and surrounding white matter are visible. We utilized an edge contrast enhancing technique (gradient filter) in order to better visualize the dentate gyrus of the hippocampus and boundaries between the hippocampus and the amygdala. The anterior boundary of the hippocampus at the junction with the amygdala is defined by the presence of the temporal horn of the lateral ventricle (lateral and superior to the genu of the hippocampus), and the posterior boundary is traced throughout its entire extent, past the level of the pulvinar, until it is no longer visible. Intertracer reliability for this study was assessed by counting the number of pixels common to two tracers’ regions of interest outlines divided by the average number of pixels in each traced region of interest. For 10 subjects, this was 86% for the amygdala and 88.3% for the hippocampus.
Identification of the ACC
Cortical Brodmann’s area measurements were used to identify the ACC (Brodmann’s areas 24, 25, 32 combined), based on the Perry coronal atlas of the brain, 49 as described in detail elsewhere. 50 – 52 This procedure used a digitized version of a detailed postmortem histologically based atlas that included 33 coronal slices of the whole brain, less the temporal lobe, and 28 coronal sections through the temporal lobe (14 in each hemisphere). Image segmentation into gray and white pixels was performed using cutoff values determined by visual examination of each subject’s axial within-brain-edge histogram. Relative glucose metabolic rate was measured from each subject’s coregistered PET and was calculated by dividing the mean FDG uptake values for the gray matter in each Brodmann area by the whole brain mean FDG uptake value. These methods provide standard and known brain atlas locations for replication.
Statistics
The overall analytic strategy was to examine group differences in response to placebo and HCORT administration (HCORT-placebo) in the hippocampus, ventral amygdala, dorsal amygdala, and ACC as the main regions of interest, using univariate and multivariate analysis of covariance (MANCOVA) followed by pair-wise comparisons for significant interactions. All regions were divided by hemisphere. Additional subdivisions in the ACC were by gray versus white matter and the three Brodmann’s areas comprising it. An additional analysis compared the effects of HCORT in the four regions of interest traced on the right and left amygdala and hippocampus.
Age was used as a covariate since it was significantly associated with the placebo rGMR (r=−0.36, df=31, p=0.045) and change in rGMR (r=0.41, df=31, p=0.020) in the dorsal amygdala. There was no significant association of hippocampal volume with hippocampal rGMR, of ventral and dorsal amygdala volume with ventral and dorsal rGMR respectively, and whole brain volume with the rGMR of the ACC. Accordingly, volumetric measures were not used as covariates. Presence or absence of comorbid depressive disorder was significantly associated only with hippocampal rGMR after controlling for age (r=0.36, df=29, p=0.046), and was used as an additional covariate.
Correlational analyses of rGMR, exploratory analysis of observed changes in laterality in the four regions, and previously reported improvement in working memory resulting from HCORT administration 9 were performed in each group separately.
RESULTS
Demographics, clinical characteristics, and volumetric measurements of the sample are summarized in Table 1 . There were no significant group differences in demographic characteristics or in the total volumes of the hippocampus, amygdala, or the whole brain. The PTSD+ group had higher rates of current and past depressive disorder and a trend toward higher rates of past alcohol dependence. The period of abstinence from alcohol abuse and dependence ranged from 2.5 to 43 years, with a mean abstinence period of 19.9 years and a median of 19.5 years.
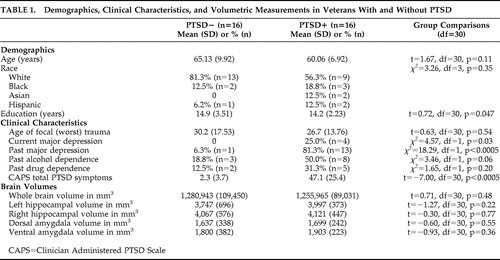 |
Hippocampus rGMR for Placebo and in Response to HCORT
Mean rGMR are presented in Figure 1 , Panel A. On the placebo day, there was a main effect of Group on hippocampal rGMR (F=4.31, df=1, 28, p=0.047) reflecting lower rGMR in the PTSD+ (0.827±0.012) than the PTSD− (0.863±0.012) group. A significant group × hemisphere interaction (F=4.87, df=1, 28, p=0.036) reflected that the group difference was only significant in the right (F=7.45, df=1, 28, p=0.011; PTSD+: 0.822±0.013, PTSD−: 0.875±0.013) but not the left (F=1.12, df=1, 28, p=0.298; PTSD+: 0.832±0.012, PTSD−: 0.852±0.012) hemisphere. The data are expressed in terms of laterality (left minus right rGMR) in Figure 1 , Panel B. Here the mean values demonstrate that in the PTSD+ group, rGMR was higher in the left than the right hippocampus; in the PTSD− group rGMR was higher in the right than the left hippocampus. However, significant hemispheric differences in rGMR were present in the PTSD− group (t=−2.09, df=15, p=0.054) but not the PTSD+ group (t=0.24,, df=15, p=0.81), as assessed by paired sample t tests.
A: Data represents the adjusted mean rGMR values (corrected for age) in each hemisphere after placebo and HCORT administration. The PTSD+ group (n=16) is represented by filled circles, and the PTSD− group (n=16) is represented by open circles. The mean±SE values are represented by SE bars.
B: Data from Panel A are redrawn from the perspective of laterality, as defined by left rGMR minus right rGMR, the placebo and HCORT days. The zero line represents no left versus right differences. Negative numbers reflect greater rGMR on the right and positive numbers reflect greater rGMR on the left.
When the analysis was repeated adding drug as a main effect, there was a significant drug × group interaction (F=4.63, df=1, 28, p=0.04) and a drug × group × hemisphere interaction (F=4.23, df=1, 28, p=0.049). This reflects that there was a significant drug by hemisphere interaction in the PTSD+ group (F=5.40, df=1, 14, p=0.04) but not in the PTSD− group (F=2.08, df=1, 14, p=0.17), controlling for age. Figure 1 , Panel A demonstrates that the effect of HCORT was to increase rGMR in the PTSD+ group but decrease rGMR in the PTSD− group. The effect in the PTSD+ group was greater in the right hemisphere. This had the result of eliminating group differences in laterality ( Figure 1 , Panel B).
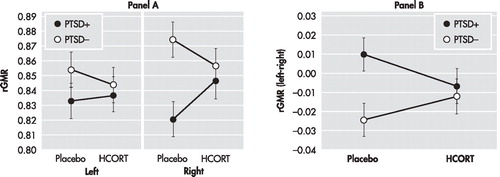
Ventral and Dorsal Amygdala rGMR at Placebo and in Response to HCORT
Ventral Amygdala
In the absence of an overall main effect of group (F=0.75, df=1, 29, p=0.39), there was a significant group × hemisphere interaction (F=4.94, df=1, 29, p=0.034) in rGMR of the ventral amygdala in the placebo condition, as shown in Figure 2 , Panel A, resulting in significant group differences in laterality (Panel B). Post-hoc testing confirmed that, as with the hippocampus, trend-level higher rGMR in the right than left hemisphere (t=−2.10, df=15, p=0.053) was only present in the PTSD− group, whereas no significant hemispheric differences in rGMR were observed in the PTSD+ group (t=0.87, df=15, p=0.40).
A: Data represents the adjusted mean rGMR values (corrected for age) in each hemisphere after placebo and HCORT administration. The PTSD+ group (n=16) is represented by filled circles, and the PTSD− group (n=16) is represented by open circles. The mean±SE values are represented by SE bars.
B: Data from Panel A are redrawn from the perspective of laterality, as defined by left rGMR minus right rGMR, the placebo and HCORT days. The zero line represents no left versus right differences. Negative numbers reflect greater rGMR on the right and positive numbers reflect greater rGMR on the left.
Here too, the effect of HCORT was to reduce the discrepancy between groups in rGMR observed on the placebo day, and to eliminate group differences in laterality, as suggested by a significant drug × group × hemisphere interaction (F=4.23, df=1, 29, p=0.049). Figure 2 , Panel A, demonstrates that rGMR increased in both groups in both hemispheres in response to HCORT, but post hoc testing demonstrated that the increase was significant only in the left hemisphere in the PTSD− group (t=−2.49, df=15, p=0.025), but not in the right hemisphere (t=−0.61, df=15, p=0.550); for the PTSD+ groups, the increases were not significant in either hemisphere (left: t=−0.15, df=15, p=0.880; right: t=1.09, df=15, p=0.292). As a result, laterality scores following HCORT no longer differed in the two groups ( Figure 2 , Panel B).
Dorsal Amygdala
There were no significant main effects or interaction of group and hemisphere in the placebo condition or following treatment with HCORT. The data are graphed in Figure 2 to demonstrate that while significant effects were not observed, the patterns in the dorsal amygdala were similar to those in the ventral amygdala
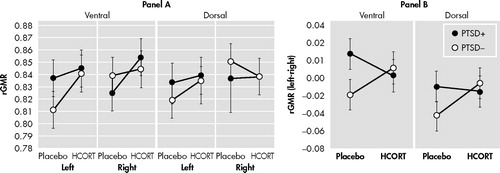
ACC in Response to Placebo and HCORT
The analyses of the ACC were conducted in a similar manner but included an additional main effect of matter (which was highly significant (F=43.73, df=1, 29, p<0.0005), reflecting greater activity in the gray than white matter, and did not differ by group (F=2.15, df=1, 29, p=0.15). A main effect of Brodmann’s area was also included so that subdivisions of the ACC based on the three Brodmann’s area from which it was comprised could be more closely examined. No main effect of Brodmann’s area or group × Brodmann’s area effects were noted.
The only significant effect of HCORT was a drug × group × matter interaction (F=8.48, df=1, 29, p=0.007). This interaction reflected that the effects of HCORT were in different directions in the two groups in the gray matter ( Figure 3 , Panel A), but post hoc testing did not confirm significance within group effects.

A: In the ACC, there was a drug × group × matter interaction showing that the effect of HCORT was confined to the gray matter, shown in Panel A. In the PTSD+ group rGMR decreased from 1.059±0.012 to 1.038±0.014, while the PTSD− group increased from 1.041±0.12 to 1.051±0.014.
B: Panel B shows the delta rGMR (HCORT – placebo) for hippocampus, amygdala, and gray matter of the anterior cingulate gyrus. The drug effect was most pronounced in the ACC (F=4.076, df=1, 28, p=0.053).
Figure 3 , Panel B demonstrates that when the magnitude of change (rGMR HCORT−rGMR placebo) was examined in all four regions (hippocampus, ventral amygdala, dorsal amygdala, ACC) in a three-way MANCOVA, there was a group × region interaction (F=4.08, df=1, 28, p=0.053). The regions of the limbic system showed quite different patterns of response to HCORT, with PTSD+ subjects showing increases in both the hippocampus and ventral amygdala, but decreases in the ACC. In contrast PTSD− subjects showed decreases in the hippocampus, increases in the ACC, and little change in the amygdala.
Correlations Between the Amygdala and ACC
On the placebo day a significant inverse relationship between the total amygdala and ACC rGMR was found in the PTSD− group (r=−0.55, df=14, p=0.028) ( Figure 4 , Panel A), reflecting strong negative relationships with the dorsal (r=−0.56, df=14, p=0.025) and the ventral (r=−0.42, df=14, p=0.10) regions (not graphed separately). A similar association between the amygdala and ACC was not present in the PTSD+ group (r=0.28, df=14, p=0.29), and tended toward the opposite direction [though it was not significantly different from the PTSD- group using Fisher’s Z (z=−0.84, df=14, p=0.40)]. Following HCORT, the two groups showed similar (though nonsignificant) correlations [PTSD− group (r=−0.15, df=14, p=0.58), PTSD+ group (r=−0.19, df=14, p=0.49)], ( Figure 4 , Panel B).

In both Panel A and B the PTSD+ group (n=16) is represented by filled circles, and the comparison group (n=16) by open circles. For Panel A the solid line shows the trend line for the PTSD+ group, r=0.277, and the dotted line shows the trend line for the comparison group, r=−0.547. For Panel B, the solid line shows the trend line for the PTSD+ group, r=−0.187, and the dotted line shows the trend line for the comparison group, r=−0.149 (both ns)
Correlations Between Memory and rGMR Changes in Response to HCORT
Given our previous finding of a significant improvement in working memory in PTSD+ following HCORT compared to performance on the placebo day, compared to effects in the PTSD− group (which were comparable on both days), 9 we examined the relationship between change in memory performance and rGMR and change in rGMR and laterality in each region of interest. The only significant correlation (r=−0.53, df=30, p=0.002) was between change in working memory and change in laterality of the dorsal amygdala ( Figure 5 ). This remained significant after adjusting the significance level by the Bonferroni inequality for multiple correlations, and was further enhanced by a partial correlation procedure controlling for age, group, and the other seven changes in rGMR and laterality (r=−0.64, df=21, p=0.001).
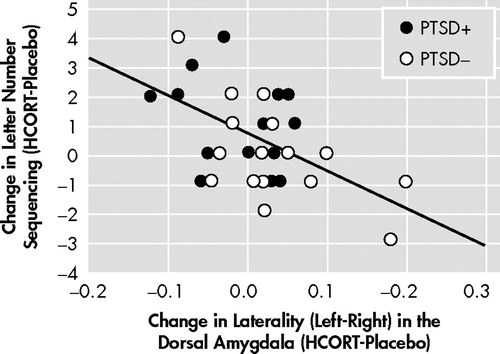
The PTSD+ group (n=16) is represented by filled circles, and the comparison group (n=16) by open circles. The solid line shows the trend line for the relationship between the change in letter number sequencing scores from placebo to HCORT days and the change in the laterality (left-right) in the dorsal amygdala, r=−0.527.
DISCUSSION
The results demonstrate regional differences in response to HCORT. The PTSD+ group showed a decrease in activity in the ACC with HCORT while the PTSD− group showed an increase, which reflects differences in central glucocorticoid responsiveness. The effect of HCORT was more prominent in the gray than white matter, consistent with a greater concentration of glucocorticoid responsiveness in gray than white matter in the human prefrontal cortex. 53 However, the pattern of decreased rGMR following HCORT was not recapitulated in the hippocampus and amygdala. Compared to the PTSD− group, the PTSD+ group responded to HCORT by increasing metabolism in the right hippocampus and the right ventral amygdala.
The effect of HCORT administration in the PTSD− group is consistent with findings in similarly aged healthy adults who also showed decreased hippocampal metabolism without effect in the prefrontal cortex following injection of an even larger dose of HCORT. 39 The negligible response to HCORT in the ACC in the PTSD− group is compatible with the demonstration of a relatively modest effect on rGMR in the ACC in combat comparison subjects following exposure to an olfactory stimulus designed to trigger traumatic memory compared to a neutral olfactory condition. 25 Yet, in the PTSD+ group, this exposure resulted in increased rGMR in the ACC compared to the neutral condition. Some recent studies have demonstrated increased activity in the medial prefrontal cortex/ACC in PTSD in response to a script-driven provocation paradigm, 26 and following nonconscious processing of fear. 27 However, the majority of reports demonstrate decreased activity in response to provocation, 20 – 23 , 28 leading to the hypothesis that the medial prefrontal cortex is hyporesponsive in PTSD. Similarly, many studies in PTSD report increased brain activity in the amygdala in response to provocation, 13 , 22 – 23 , 25 as observed in the ventral amygdala in the current study in response to HCORT. Together, the findings of decreased activity in the medial prefrontal cortex/ACC and increased activity in the amygdala during various conditions in PTSD lead to the hypothesis that disruptions in the normal neurocircuitry of these regions manifest as a failed containment of amygdala activity by the medial prefrontal cortex/ACC. 39
In the present study, stronger effects were detected in the ACC than hippocampus or amygdala, possibly attributed to a greater concentration of glucocorticoid responsiveness in the prefrontal cortex. 54 Interestingly, regardless of the extent or direction of group differences in rGMR on the placebo day, the effect of HCORT, in all cases, was to reduce the differences observed on the placebo day. In the hippocampus and amygdala, this was manifest by decreasing group differences in hemispheric laterality, whereas in the ACC this was demonstrated by a reduction in rGMR in the PTSD+ group.
The current findings showing hemispheric differences may reflect the fact that the right hemisphere operates with a greater range of activity—thus, the addition of HCORT would be greater due to increased variation. However, differences in hemispheric laterality may underlie differences observed regarding right and left hemispheric differences in provocation-related activation patterns, in association with ACC activity, in the previously published literature. To date, almost every study examining the hippocampus, amygdala, or ACC has found effects to be restricted to one hemisphere, or directionally different in the two hemispheres. 17 , 19 , 21 , 22 That there is an association between brain laterality and PTSD risk has recently been confirmed in a large epidemiologic study demonstrating mixed lateral preferences for handedness was associated with PTSD. 55 Differences in handedness do not explain the current findings because only one subject in the sample was left handed. However, the original hypothesis regarding brain laterality emphasized that those with a reduced cerebral lateralization may be less able to detect and/or engage in corrective cognitive processing of threatening stimuli. 56 In the current study, there was less laterality in hippocampal and amygdala rGMR in PTSD on the placebo day, and HCORT reduced these group differences by increasing lateralization in the PTSD+ group and decreasing it in the PTSD− group. Resting data with 18 FDG-PET can assess trait-like laterality differences present without a specific psychological stimulus. Blood-oxygen-level-dependent (BOLD) activation studies are only able to interpret laterality (or hemispheric) differences in connectivity between PTSD and comparison groups in response to stimulation. Yet, numerous such observations have been made, supporting the importance of laterality differences in brain function in PTSD.
Interestingly, the only region to demonstrate an activation-related association with improvement in working memory was the dorsal amygdala. Though group differences for this region did not approach statistical significance, the correlation between brain activity in this region and memory performance is compatible with results of animal studies demonstrating that glucocorticoid induced effects on memory are enabled by noradrenergic activation of the basolateral amygdala. 57
The findings add to the literature not only in their demonstration of group differences in the effects of HCORT, but also in their description of group differences in rGMR under resting conditions without pharmacological challenge. The differences on the placebo day, in particular, provide a context for interpreting data in response to provocation. Previously reported group differences in the direction of amygdala and/or ACC activity in response to provocation may be misleading without consideration of baseline activation. Had our study simply examined metabolic effects of HCORT administration on PTSD+ compared to PTSD− we would have likely concluded that effects were minimal, if not absent.
The negative correlation between ACC and amygdala metabolism observed on the placebo day in the PTSD− group implies that this neural circuit may regulate amygdala activity even under ambient conditions. 58 – 60 In the present study, this relationship was absent, and even in the opposite direction (although nonsignificant), in subjects with PTSD. Furthermore, in PTSD the rGMR in the ACC under placebo conditions was increased relative to comparison subjects. It is difficult to interpret the effect of this activity on the amygdala since right and left hemispheres showed different responses in PTSD. In that the PTSD+ group on the placebo day showed greater activity of the amygdala in the left, but lower activity in the right compared to the PTSD− group, the placebo ACC/amygdala relationship can either be interpreted as inverse or positive depending on the hemisphere. In response to a single dose of HCORT, the associations between ACC and amygdala metabolism were no longer discrepant between the PTSD+ and PTSD− groups, nor were differences in these associations present in the two hemispheres. It is particularly noteworthy that the effect of HCORT resulted in similar associations between amygdala and ACC activity in both groups on the active treatment day since the effect of HCORT in PTSD+ subjects was to restore a normal inverse association, while PTSD− subjects experienced a disruption of this normal neural network. A limitation of the study is that without a healthy comparison group not exposed to combat it is unclear whether the PTSD− group also represents a group with altered glucocorticoid function. Additionally, as we only studied male veterans, these results may not be generalizable.
Regarding possible treatment implications, the beneficial effects of glucocorticoids have been investigated and continue to be of interest, particularly in PTSD prophylaxis. 3 , 61 The current observations are consistent with the beneficial effects noted on memory observed in this older cohort. However, in a younger cohort HCORT resulted in impairment of working memory, 8 consistent with a larger literature in healthy subjects. 62 – 63 Regionally specific differential responses to glucocorticoids have been noted according to age, 64 – 69 and their relevance should be fully explored since it may be that therapeutic approaches that are optimal at some phases of illness are contraindicated in others based on age, chronicity, or other factors contributing to dynamic changes in pathophysiology and brain function. Little is known about the efficacy of standard treatments of PTSD in the elderly. However the current discrepancies between aging PTSD and normal aging profiles suggests that we cannot extrapolate on the basis of the literature on normal aging to what might be true as persons with PTSD advance in age.
1. Yehuda R: Advances in understanding neuroendocrine alterations in PTSD and their therapeutic implications. Ann N Y Acad Sci 2006; 1071:137–166Google Scholar
2. Aerni A, Traber R, Hock C, et al: Low-dose cortisol for symptoms of posttraumatic stress disorder. Am J Psychiatry 2004; 161:1488–1490Google Scholar
3. Schelling G, Kilger E, Roozendaal B, et al: Stress doses of hydrocortisone, traumatic memories, and symptoms of posttraumatic stress disorder in patients after cardiac surgery: a randomized study. Biol Psychiatry 2004; 55:627–633Google Scholar
4. Schelling G, Roozendaal B, de Quervain DJ: Can posttraumatic stress disorder be prevented with glucocorticoids? Ann N Y Acad Sci 2004; 1032:158–166Google Scholar
5. Sher L, Oquendo MA, Li S, et al: Higher cerebrospinal fluid homovanillic acid levels in depressed patients with comorbid posttraumatic stress disorder. Eur Neuropsychopharmacol 2005; 15:203–209Google Scholar
6. Geracioti TD Jr, Baker DG, Ekhator NN, et al: CSF norepinephrine concentrations in posttraumatic stress disorder. Am J Psychiatry 2001; 158:1227–1230Google Scholar
7. Baker DG, West SA, Nicholson WE, et al: Serial CSF corticotropin-releasing hormone levels and adrenocortical activity in combat veterans with posttraumatic stress disorder. Am J Psychiatry 1999; 156:585–588Google Scholar
8. Grossman R, Yehuda R, Golier J, et al: Cognitive effects of intravenous hydrocortisone in subjects with PTSD and healthy control subjects. Ann N Y Acad Sci 2006; 1071:410–421Google Scholar
9. Yehuda R, Harvey PD, Buchsbaum M, et al: Enhanced effects of cortisol administration on episodic and working memory in aging veterans with PTSD. Neuropsychopharmacology 2007; 32:2581–2591Google Scholar
10. Perlman WR, Webster MJ, Herman MM, et al: Age-related differences in glucocorticoid receptor mRNA levels in the human brain. Neurobiol Aging 2007; 28:447–458Google Scholar
11. Bizon JL, Helm KA, Han JS, et al: Hypothalamic-pituitary-adrenal axis function and corticosterone receptor expression in behaviourally characterized young and aged long-evans rats. Eur J Neurosci 2001; 14:1739–1751Google Scholar
12. Yau JL, Olsson T, Morris RG, et al: Glucocorticoids, hippocampal corticosteroid receptor gene expression and antidepressant treatment: relationship with spatial learning in young and aged rats. Neuroscience 1995; 66:571–581Google Scholar
13. Shin LM, Rauch SL, Pitman RK: Amygdala, medial prefrontal cortex, and hippocampal function in PTSD. Ann N Y Acad Sci 2006; 1071:67–79Google Scholar
14. Liberzon I, Britton JC, Phan KL: Neural correlates of traumatic recall in posttraumatic stress disorder. Stress 2003; 6:151–156Google Scholar
15. Rauch SL, Shin LM, Phelps EA: Neurocircuitry models of posttraumatic stress disorder and extinction: human neuroimaging research–past, present, and future. Biol Psychiatry 2006; 60:376–382Google Scholar
16. Lanius RA, Bluhm R, Lanius U, et al: A review of neuroimaging studies in PTSD: heterogeneity of response to symptom provocation. J Psychiatr Res 2006; 40:709–729Google Scholar
17. McEwen BS: Glucocorticoids, depression, and mood disorders: structural remodeling in the brain. Metabolism 2005; 54:20–23Google Scholar
18. Kim JJ, Haller J: Glucocorticoid hyper- and hypofunction: stress effects on cognition and aggression. Ann N Y Acad Sci 2007; 1113:291–303Google Scholar
19. Roozendaal B, McReynolds JR, McGaugh JL: The basolateral amygdala interacts with the medial prefrontal cortex in regulating glucocorticoid effects on working memory impairment. J Neurosci 2004; 24:1385–1392Google Scholar
20. Shin LM, Orr SP, Carson MA, et al: Regional cerebral blood flow in the amygdala and medial prefrontal cortex during traumatic imagery in male and female Vietnam veterans with PTSD. Arch Gen Psychiatry 2004; 61:168–176Google Scholar
21. Shin LM, Wright CI, Cannistraro PA, et al: A functional magnetic resonance imaging study of amygdala and medial prefrontal cortex responses to overtly presented fearful faces in posttraumatic stress disorder. Arch Gen Psychiatry 2005; 62:273–281Google Scholar
22. Williams LM, Kemp AH, Felmingham K, et al: Trauma modulates amygdala and medial prefrontal responses to consciously attended fear. Neuroimage 2006; 29:347–357Google Scholar
23. Bremner JD, Vermetten E, Schmahl C, et al: Positron emission tomographic imaging of neural correlates of a fear acquisition and extinction paradigm in women with childhood sexual-abuse-related post-traumatic stress disorder. Psychol Med 2005; 35:791–806Google Scholar
24. Phan KL, Britton JC, Taylor SF, et al: Corticolimbic blood flow during nontraumatic emotional processing in posttraumatic stress disorder. Arch Gen Psychiatry 2006; 63:184–192Google Scholar
25. Vermetten E, Schmahl C, Southwick SM, et al: Positron tomographic emission study of olfactory induced emotional recall in veterans with and without combat-related posttraumatic stress disorder. Psychopharmacol Bull 2007; 40:8–30Google Scholar
26. Lanius RA, Williamson PC, Boksman K, et al: Brain activation during script-driven imagery induced dissociative responses in PTSD: a functional magnetic resonance imaging investigation. Biol Psychiatry 2002; 52:305–311Google Scholar
27. Bryant RA, Kemp AH, Felmingham KL, et al: Enhanced amygdala and medial prefrontal activation during nonconscious processing of fear in posttraumatic stress disorder: an fMRI study. Hum Brain Mapp 2008; 29:517–523Google Scholar
28. Britton JC, Phan KL, Taylor SF, et al: Corticolimbic blood flow in posttraumatic stress disorder during script-driven imagery. Biol Psychiatry 2005; 57:832–840Google Scholar
29. Gilboa A, Shalev AY, Laor L, et al: Functional connectivity of the prefrontal cortex and the amygdala in posttraumatic stress disorder. Biol Psychiatry 2004; 55:263–272Google Scholar
30. Kim H, Somerville LH, Johnstone T, et al: Inverse amygdala and medial prefrontal cortex responses to surprised faces. Neuroreport 2003; 14:2317–2322Google Scholar
31. Nomura M, Ohira H, Haneda K, et al: Functional association of the amygdala and ventral prefrontal cortex during cognitive evaluation of facial expressions primed by masked angry faces: an event-related fMRI study. Neuroimage 2004; 21:352–363Google Scholar
32. Smith AP, Stephan KE, Rugg MD, et al: Task and content modulate amygdala-hippocampal connectivity in emotional retrieval. Neuron 2006; 49:631–638Google Scholar
33. Taylor SF, Liberzon I, Fig LM, et al: The effect of emotional content on visual recognition memory: a PET activation study. Neuroimage 1998; 8:188–197Google Scholar
34. Whalen PJ, Shin LM, McInerney SC, et al: A functional MRI study of human amygdala responses to facial expressions of fear versus anger. Emotion 2001; 1:70–83Google Scholar
35. Urry HL, van Reekum CM, Johnstone T, et al: Amygdala and ventromedial prefrontal cortex are inversely coupled during regulation of negative affect and predict the diurnal pattern of cortisol secretion among older adults. J Neurosci 2006; 26:4415–4425Google Scholar
36. Bremner JD, Vythilingam M, Vermetten E, et al: Neural correlates of declarative memory for emotionally valenced words in women with posttraumatic stress disorder related to early childhood sexual abuse. Biol Psychiatry 2003; 53:879–889Google Scholar
37. Lanius RA, Frewen PA, Girotti M, et al: Neural correlates of trauma script-imagery in posttraumatic stress disorder with and without comorbid major depression: a functional MRI investigation. Psychiatry Res 2007; 155:45–56Google Scholar
38. Yehuda R, Yang RK, Buchsbaum MS, et al: Alterations in cortisol negative feedback inhibition as examined using the ACTH response to cortisol administration in PTSD. Psychoneuroendocrinology 2006; 31:447–451Google Scholar
39. de Leon MJ, McRae T, Rusinek H, et al: Cortisol reduces hippocampal glucose metabolism in normal elderly, but not in Alzheimer’s disease. J Clin Endocrinol Metab 1997; 82:3251–3259Google Scholar
40. Golier JA, Harvey PD, Legge J, et al: Memory performance in older trauma survivors: implications for the longitudinal course of PTSD. Ann NY Acad Sci 2006; 1071:54–66Google Scholar
41. Gazdzinski S, Durazzo TC, Meyerhoff DJ: Temporal dynamics and determinants of whole brain tissue volume changes during recovery from alcohol dependence. Drug Alcohol Depend 2005; 78:263–273Google Scholar
42. Pfefferbaum A, Sullivan EV, Mathalon DH, et al: Longitudinal changes in magnetic resonance imaging brain volumes in abstinent and relapsed alcoholics. Alcohol Clin Exp Res 1995; 19:1177–1191Google Scholar
43. Spitzer RL, Williams JBW, Gibbon M: Structured Clinical Interview for DSM-IV (SCID). New York, New York State Psychiatric Institute, Biomedical Research, 1995Google Scholar
44. Blake DD, Weathers FW, Nagy LM, et al: The development of a clinician-administered PTSD scale. J Trauma Stress 1995; 8:75–90Google Scholar
45. Hazlett EA, Buchsbaum MS, Mohs RC, et al: Age-related shift in brain region activity during successful memory performance. Neurobiol Aging 1998; 19:437–445Google Scholar
46. Jenkinson M, Smith S: A global optimisation method for robust affine registration of brain images. Med Image Anal 2001; 5:143–156Google Scholar
47. Yehuda R, Golier JA, Tischler L, et al: Hippocampal volume in aging combat veterans with and without post-traumatic stress disorder: relation to risk and resilience factors. J Psychiatr Res 2007; 41:435–445Google Scholar
48. Haznedar MM, Buchsbaum MS, Wei TC, et al: Limbic circuitry in patients with autism spectrum disorders studied with positron emission tomography and magnetic resonance imaging. Am J Psychiatry 2000; 57:1994–2001Google Scholar
49. Perry RH: Personal communication, 1993Google Scholar
50. Buchsbaum MS, Nenadic I, Hazlett EA, et al: Differential metabolic rates in prefrontal and temporal brodmann areas in schizophrenia and schizotypal personality disorder. Schizophr Res 2002; 54:141–150Google Scholar
51. Stein DJ, Buchsbaum MS, Hof PR, et al: Greater metabolic rate decreases in hippocampal formation and proisocortex than in neocortex in Alzheimer’s disease. Neuropsychobiology 1998; 37:10–19Google Scholar
52. Mitelman SA, Buchsbaum MS, Brickman AM, et al: Cortical intercorrelations of frontal area volumes in schizophrenia. Neuroimage 2005; 27:753–770Google Scholar
53. Sanchez MM, Young LJ, Plotsky PM, et al: Distribution of corticosteroid receptors in the rhesus brain: relative absence of glucocorticoid receptors in the hippocampal formation. J Neurosci 2000; 20:4657–4668Google Scholar
54. Boscarino JA, Hoffman SN: Consistent association between mixed lateral preference and PTSD: confirmation among a national study of 2490 US army Vietnam veterans. Psychosom Med 2007; 69:365–369Google Scholar
55. Chemtob CM, Wang Y, Dugan KL, et al: Mixed lateral preference and peritraumatic reactions to the World Trade Center attacks. J Nerv Ment Dis 2006; 194:874–876Google Scholar
56. Roozendaal B, Hahn EL, Nathan SV, et al: Glucocorticoid effects on memory retrieval require concurrent noradrenergic activity in the hippocampus and basolateral amygdala. J Neurosci 2004; 24:8161–8169Google Scholar
57. Das P, Kemp AH, Liddell BJ, et al: Pathways for fear perception: modulation of amygdala activity by thalamo-cortical systems. Neuroimage 2005; 26:141–148Google Scholar
58. Likhtik E, Pelletier JG, Paz R, et al: Prefrontal control of the amygdala. J Neurosci 2005; 25:7429–7437Google Scholar
59. Schelling G, Roozendaal B, Krauseneck T, et al: Efficacy of hydrocortisone in preventing posttraumatic stress disorder following critical illness and major surgery. Ann N Y Acad Sci 2006; 1071:46–53Google Scholar
60. New AS, Hazlett EA, Buchsbaum MS, et al: Amygdala-prefrontal disconnection in borderline personality disorder. Neuropsychopharm 2007; 32:1629–1640Google Scholar
61. Lupien SJ, Wilkinson CW, Briere S, et al: The modulatory effects of corticosteroids on cognition: studies in young human populations. Psychoneuroendocrinology 2002; 27:401–416Google Scholar
62. Newcomer JW, Selke G, Melson AK, et al: Decreased memory performance in healthy humans induced by stress-level cortisol treatment. Arch Gen Psychiatry 1999; 56:527–533Google Scholar
63. Miller DB, O’Callaghan JP: Aging, stress and the hippocampus. Ageing Res Rev 2005; 4:123–140Google Scholar
64. De Leon MJ, George AE, Golomb J, et al: Frequency of hippocampal formation atrophy in normal aging and Alzheimer’s disease. Neurobiol Aging 1997; 18:1–11Google Scholar
65. de Kloet ER: Corticosteroids, stress, and aging. Ann N Y Acad Sci 1992; 663:357–371Google Scholar
66. Sapolsky RM: Glucocorticoids, stress, and their adverse neurological effects: relevance to aging. Exp Gerontol 1999; 34:721–732Google Scholar
67. Heffelfinger AK, Newcomer JW: Glucocorticoid effects on memory function over the human life span. Dev Psychopathol 2001; 13:491–513Google Scholar
68. Lupien SJ, Wilkinson CW, Briere S, et al: Acute modulation of aged human memory by pharmacological manipulation of glucocorticoids. J Clin Endocrinol Metab 2002; 87:3798–3807Google Scholar
69. Murphy EK, Spencer RL, Sipe KJ, et al: Decrements in nuclear glucocorticoid receptor (GR) protein levels and DNA binding in aged rat hippocampus. Endocrinology 2002; 143:1362–1370Google Scholar

