
A Rare Dementing Disease: Adult Neuronal Ceroid Lipofuscinoses
Abstract
To emphasize the importance of clinical and ultrastructural findings for diagnosing adult neuronal ceroid lipofuscinosis (ANCL), the authors retrospectively reviewed six patients with biopsy-proven ANCL. In all cases, ophthalmologic examinations were normal, and electron microscopic studies demonstrated characteristic granular osmiophilic deposits within the eccrine epithelial cells. The inheritance and mechanism of ANCL remain unknown, and the diagnosis is based on clinical features and ultrastructural examination of the cerebral tissue or extracerebral accumulation of lipopigments. This study suggests that ANCL should be considered as a possible diagnosis in patients with early-onset dementia.
Neuronal ceroid lipofuscinoses (NCLs) are a group of inherited, progressive neurodegenerative diseases in children and adults characterized morphologically by accumulation of ceroid lipopigment in the lysosomes of neurons and extraneural cells.1,2 NCLs are classified into five forms according to age at onset, clinical signs, and morphologic findings. These are: 1) congenital; 2) infantile (INCL; Santauvori-Haltia disease); 3) classical late-infantile NCL (LINCL; Jansky-Bielschowsky disease); 4) juvenile NCL (JNCL; Batten’s disease or Spielmeyer-Vogt-Sjögren disease); and 5) adult type of NCL (ANCL; Kufs disease). Also, there are several variant forms, especially for JNCL, and at least eight different genes (CLN1–CLN8) have been identified at present.3 Despite different clinical presentations and ages at onset, all forms of NCLs have the same histopathological components, including autofluorescent, periodic acid-Schiff and Sudan black B-positive granules, resistant to lipid solvents in neurons and extraneuronal cells.4
ANCL is the late-onset and rarest form, comprising 1.3%–10% of all NCLs.5–8 It has sporadic and autosomal recessive forms; the latter is known as Kufs’ disease, which was described by Hugo Friedrich Kufs in 1925. Kufs disease is characterized by seizures, dementia, ataxia, extrapyramidal features such as tremors or tics, and dysarthria. The signs and symptoms typically appear around age 30, but they can develop anytime between adolescence and late adulthood. There is also an autosomal dominant form, which is known as Parry disease, described by Boehme in 1971.9,10
Among eight genes, CLN4 has been assigned for ANCL, but the CLN4 gene or genes, as related to the molecular basis of ANCL, is still unknown. However, mutations in the CLN1 or PPT1 gene, although commonly associated with INCL, may also be a rare cause of ANCL and many missense mutations, some of which are reported to be associated with ANCL.11–14 In addition to CLN1, it has just been reported that mutations in the CLN6 gene, accounting for variant late-infantile NCL, and including a mixed type of fingerprint and curvilinear profiles and rectilinear complexes, is also found in some families affected by ANCL and is specified as the major cause of recessive ANCL.15
The diagnosis of ANCL disease is still based on clinical features and morphological examination of the cerebral tissue or extracerebral accumulation of lipopigments. Ultrastructurally, ANCL in affected cells is usually associated with the mixed type of fingerprint (FP) and curvilinear profiles (CV), granular osmiophilic deposits (GROD), and rectilinear complexes (RC).1,2,16
In this article, we are reporting on six patients who were referred to us because of early-onset dementia with and without seizure, all of whom received the diagnosis of biopsy-proven Kufs’ disease.
Method
We reviewed a series of six patients with ANCL in a 5-year period. The evaluations consisted of family history, neurologic examination, cranial magnetic resonance imaging (MRI), neuropsychological assessment, electroencephalography, and ophthalmologic examination. All patients underwent a skin-punch biopsy for the purpose of ultrastructural examination by electron microscopy. For all patients, detailed cerebro-spinal fluid (CSF) and blood chemistry tests were evaluated. In Case 5, cerebral angiography was performed in order to exclude central nervous system vasculitis.
Results
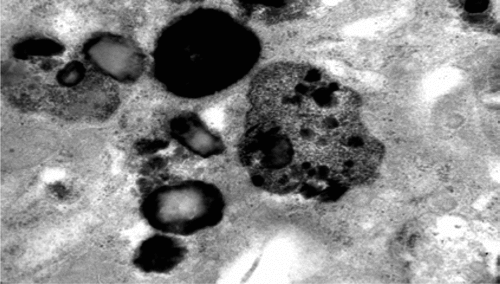
Age at onset in the series varied between 20 years and 61 years (mean: 44; standard deviation [SD]: 14.8). At the time of our first examination, the duration of disease ranged from 2 years to 10 years (mean: 6.83 [3.7]). Behavioral abnormalities with psychotic features were the most frequent initial symptoms, and dementia was the most common presenting symptom. With the exception of Case 4, all of the patients had seizures. None of them had visual failure during the course of the illness, and ophthalmologic examinations were normal. Ultrastructural examination of a cutaneous biopsy, performed by electron microscopy showed granular osmophilic deposits in eccrine sweat gland epithelial cells (Figure 1, Figure 2).

In all cases, MRI studies revealed varying degrees of cerebral atrophy, with an increase in T2-weighted signal in periventricular distribution and ventricular enlargement (Figure 3).
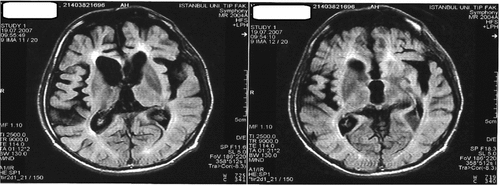
In Case 5, signal abnormalities were also present in the immediate subcortical areas, which were suggestive of cerebral vasculitis. However, cerebral angiogram and vasculitis markers were normal, and a repeat CSF 14-3-3 protein test did not confirm the previous positive result, which had been done elsewhere.
Only two patients had dementia in their family history; since they did not agree to any examination, we could not decide these family members’ diagnosis.
On interictal EEG recordings, slowing of background activity was noted in all cases.
Neuropsychological tests showed varying degrees of cognitive deterioration in all areas. In Case 5, neuropsychological assessment could not be done because of the patient’s severe cognitive decline.
Clinical findings are summarized in Table 1.
| Case #1 | Case #2 | Case #3 | Case #4 | Case #5 | Case #6 | |
|---|---|---|---|---|---|---|
| Sex | M | M | M | M | F | F |
| Age at onset, years | 51 | 34 | 20 | 45 | 53 | 61 |
| Current age, tears | 53 | 44 | 30 | 51 | 56 | 71 |
| Family history | — | — | — | + | — | + |
| Previous history | DM | unremarkable | febrile seizure | HTA | DM, HTA | hepatitis C, HTA |
| Consanguinity | — | + | — | — | — | — |
| First symptom | GTC seizure | psychotic signs | psychotic signs | psychotic signs | forgetfulness, psychotic signs | parkinsonism |
| Subsequent symptoms | forgetfulness, psychotic signs | GTC seizure | psychotic signs | forgetfulness | myoclonus, seizure | forgetfulness, psychotic signs |
| Presenting symptoms | dementia | dementia | dementia, seizure | dementia | dementia | parkinsonism, dementia, seizure |
| Years since dementia onset | 1 | 7 | 7 | 3 | 2 | 5 |
| Facial dyskinesia | — | + | — | + | — | + |
| Parkinsonism | — | + | — | + | + | + |
| Myoclonus | — | + | — | + | + | + |
| Ataxia | — | + | — | + | + | + |
| Seizures | GTC seizure | GTC seizure | GTC seizure | (no seizure) | myoclonic epilepsy | GTC seizure |
| RSBD | — | — | — | — | — | + |
| Response to seizure treatment | + | + | + | (no seizure) | + | + |
| Mini-Mental State Exam | 19 | 8 | 15 | 20 | N/A | 5 |
| Epileptiform EEG | + | + | + | + | + | + |
| MRI abnormalities | + | + | + | + | + | + |
| Retinal finding | — | — | — | — | — | — |
| Electron microscopy | GROD | GROD | GROD | GROD | GROD | GROD |
Discussion
The clinical signs of ANCL are very heterogeneous. Berkovic et al. described two types of Kufs disease, depending on the dominant clinical feature.1 Type A presents initially with generalized tonic–clonic seizures, followed by progressive myoclonic seizures, photosensitivity, dementia, ataxia, and pyramidal and extrapyramidal signs. Type B starts with behavioral disturbances and dementia, and, subsequently, motor disturbances, particularly tic-like facial dyskinesia; ataxia, extrapyramidal, and bulbar signs are added.1,2,7,17,18 Although this classification is widely accepted, atypical cases that did not fit the above clinical categories but were pathologically proven as ANCL disease have also been reported.1,19 Moreover, there may be many overlaps between the two types.20 Since pyramidal and extrapyramidal signs in type A, and seizures in type B, develop in later stages of the disease, the lack of either seizures or motor features early in the course of an early-onset dementia case should not rule out the possibility of ANCL.1 ANCL disease classically differs from other NCLs by absence of retinal degeneration.1,2 The patients described here had a similar clinical presentation, apart from Case 1, who had a generalized tonic–clonic (GTC) seizure as the first symptom. We used the criteria determined by Berkovic to categorize types and considered that five patients fit into the type B, and Case 1 fit type A.
The course of the disease is slowly progressive, and duration is 12.4 (SD: 9.4) years.1,21 In ANCL disease, clinical signs typically start around the age of 30 years, but age at onset varies between 11 and 50 years. In Case 6, age at onset (61 years) was late, as compared with the rest of our group and other cases reported previously.1,16,21
Electrophysiological and neuroradiological findings are not specific in ANCL.22,23 At the initial stages of the disease, cerebral atrophy is either ambiguous or absent, with no white-matter abnormalities.24 In later stages, cerebral and cerebellar atrophy and a diffuse white-matter changes on T2-weighted and FLAIR images were reported in the MRI studies.25–27 MRI in our cases showed nonspecific diffuse cerebral atrophy and white-matter abnormalities that were not suggestive of any other neurodegenerative dementing illness. Nonspecific EEG changes with background slowing were reported in the two types of pathologically-proven ANCL.1 The EEG recordings of all our cases indicated irregular slow background activity. For the treatment of seizures, antiepileptic drugs should be selected with caution because carbamazepine, phenytoin, and lamotrigine may increase seizures.28 Valproic acid29 and zonisamid7 may be more effective for the purpose of seizure control. Over the course of disease, seizures may become drug-resistant, and status epilepticus may develop. Seizures in our first patient were still frequent, despite high-dose levetiracetam treatment. Valproic acid add-on enabled the patient to be seizure-free. In our series, two cases were women. We are not aware of any study reporting gender difference; however, to-date, there seems to be a preponderance of women in the reported cases.
Since the molecular defect of ANCL is not understood, its diagnosis depends on the demonstration of specific histopathological findings by the ultrastructural examination, and, through these features, ANCL can be recognized and differentiated from other neurodegenerative dementias. Electron microscopic examination of skin biopsy is a simple, noninvasive, and safe method to approach this, and currently is the only way to confirm the diagnosis.1,2,4 Ultrastructurally, mixed type of inclusions can be observed, but granular profile is the most frequently reported pattern.2 In terms of storage material, no differences were reported between the two types.1 However, GROD have been observed in autosomal dominant forms of ANCL.7,13 There is an autosomal recessive mode of inheritance in most cases; however, very few patients with autosomal dominant inheritance (Parry type) have been reported in literature.7,13,22 All patients in this series had GROD-type inclusion; however, only in two cases was there dementia in the family history. Because of absence of family history, different type of accumulation of lipopigments apart from GROD was expected to be seen in four other cases. It is important to keep in mind that mutations in CLN1/PPT1 may be underlying in such patients. If characteristic GROD inclusions are present, PPT1 enzyme activity should be assayed to ensure an accurate diagnosis. Unfortunately, in these cases, neither enzyme activity evaluation nor the molecular analysis for genetic sequencing could be performed for CLN1/PPT1. Mutations in the CLN6 gene were not considered because of different type lipopigments. Skin biopsy is necessary for the definitive diagnosis of ANCL, but the pathologic specimen may be inadequate for the inclusions to be visualized, and a repeat biopsy may become necessary.
The delay in the diagnosis might have several reasons. One likely reason is that the first referral is frequently made to a psychiatrist because of the initial presentation with psychosis and unresponsiveness to treatment. The addition of neurologic findings prompts these patients to be referred to a neurologist. Five of our six patients had been treated by a psychiatrist initially because of long-standing psychosis before being sent to us after cognitive decline and/or seizures became evident. Since our patients did not have any psychological testing related to psychotic signs, our evaluations are based on the history only from the patients’ relatives. According to the information given by relatives of patients, aggressive, agitated behaviors, personality changes, and then delusional ideation were more likely to be reported as psychotic signs, and the onset of symptoms was insidious. Delusional jealousy and persecutory delusions were prominent. Delusions were not persistent and systematized, and there was some fluctuation in intensity and frequency. Hallucinations were not early psychotic signs, but occurred later and were usually visual. There was a poor response to treatment with antipsychotics. Memory problems, word-finding, and praxic abilities were not an early issue, nor was a history of attempted suicide or suicidal ideation or disorganized behavior and speech.
The common dementing diseases, that is, Alzheimer’s disease, fronto-temporal dementia, and dementia associated with Lewy bodies can be more or less easily differentiated on the basis of specific clinic criteria, neuropsychological profile, and neuroradiological findings. However, the current level of understanding of a rare disease such as ANCL does not facilitate clinicians in arriving at a straightforward differential diagnosis. Unlike most of the other neurodegenerative dementias, ANCL is a unique disease in which the diagnosis can be confirmed with a very easy skin biopsy. Therefore, it should be considered in patients presenting with an early-onset dementia with or without seizures and motor abnormalities and in those presenting with late-onset seizures with no obvious reason such as a mass lesion of the brain. Thus, although there is currently no available treatment, appropriate genetic counseling and support can be provided, and data from these patients point out a new approach in ANCL.
Ultrastructural finding, presence of GRODs, and lack of consanguinity, except in one case, suggest an autosomal dominant (AD) inheritance for majority of the cases. This is an important finding, since AD inheritance is reportedly the rarest cause of ANCL. Lack of genetic analysis is the obvious weakness of our series. For this reason, we do not claim a homogenous inheritance pattern with the evidence we have so far. However, in order to detect this probably-underdiagnosed condition, the six case series recruited in a relatively short time period highlights the importance of skin biopsy in early-onset dementia with or without seizures.
1 : Kufs’ disease: a critical reappraisal. Brain 1988; 111:27–62Crossref, Medline, Google Scholar
2 : Adult neuronal ceroid-lipofuscinosis. Clin Neuropathol 1989; 8:109–119Medline, Google Scholar
3 : Mutations in CLN7/MFSD8 are a common cause of variant late-infantile neuronal ceroid lipofuscinosis. Brain 2009; 132:810–819Crossref, Medline, Google Scholar
4 : The neuronal ceroid-lipofuscinoses: from past to present. Biochim Biophys Acta 2006; 1762:850–856 [Review]Crossref, Medline, Google Scholar
5 : Adult type of neuronal ceroid lipofuscinosis. Dev Neurosci 1991; 13:331–338Crossref, Medline, Google Scholar
6 : Familial Kufs’ disease presenting as a progressive myoclonic epilepsy. J Neurol 2000; 247:447–454Crossref, Medline, Google Scholar
7 : Adult-onset neuronal ceroid lipofuscinosis (Kufs disease) with autosomal dominant inheritance in Alabama. Epilepsia 2003; 44:841–846Crossref, Medline, Google Scholar
8 : Adult-onset neuronal ceroid lipofuscinosis type B in an African-American. Mov Disord 2005; 20:752–754Crossref, Medline, Google Scholar
9 : A dominant form of neuronal ceroid-lipofuscinosis. Brain 1971; 94:745–760Crossref, Medline, Google Scholar
10 : A golgi and ultrastructural study of a dominant form of Kufs’ disease. J Neurol 1980; 222:183–190Crossref, Medline, Google Scholar
11 : Adult neuronal ceroid lipofuscinosis with palmitoyl-protein thioesterase deficiency: first adult-onset patients of a childhood disease. Ann Neurol 2001; 50:269–272Crossref, Medline, Google Scholar
12 : Biochemical analysis of mutations in palmitoyl-protein thioesterase causing infantile and late-onset forms of neuronal ceroid lipofuscinosis. Hum Mol Genet 2001; 10:1431–1439Crossref, Medline, Google Scholar
13 : Autosomal dominant adult neuronal ceroid lipofuscinosis: a novel form of NCL with granular osmiophilic deposits without palmitoyl protein thioesterase 1 deficiency. Brain Pathol 2003; 13:574–581Crossref, Medline, Google Scholar
14 : Neuronal ceroid lipofuscinoses. Biochim Biophys Acta 2009; 1793:697–709Crossref, Medline, Google Scholar
15 : Kufs disease: the major adult form of neuronal ceroid lipofuscinosis, caused by mutations in CLN6. Am J Hum Genet 2011; 88:566–573Crossref, Medline, Google Scholar
16 : Neuronal ceroid lipofuscinoses: classification and diagnosis. Adv Genet 2001; 45:1–34Crossref, Medline, Google Scholar
17 : Autosomal dominant adult neuronal ceroid lipofuscinosis: parkinsonism due to both striatal and nigral dysfunction. Mov Disord 2002; 17:482–487Crossref, Medline, Google Scholar
18 : Early-onset dementia with prolonged occipital seizures: an atypical case of Kufs disease. Neurology 2008; 71:1709–1712Crossref, Medline, Google Scholar
19 : Classification of the neuronal ceroid-lipofuscinoses: expansion of the atypical forms. Am J Med Genet 1995; 57:150–154Crossref, Medline, Google Scholar
20 : Kufs’ disease presenting as progressive dementia with late-onset generalized seizures: a clinicopathological and electrophysiological study. Epilepsia 1992; 33:65–74Crossref, Medline, Google Scholar
21 : The neuronal ceroid-lipofuscinoses. J Neuropathol Exp Neurol 2003; 62:1–13Crossref, Medline, Google Scholar
22 : Autosomal dominant Kufs’ disease: a cause of early-onset dementia. J Neurol Sci 2001; 188:51–60Crossref, Medline, Google Scholar
23 : Electroencephalographic findings in Kufs disease. Clin Neurophysiol 2003; 114:1738–1743Crossref, Medline, Google Scholar
24 : MRI in neuronal ceroid lipofuscinosis. Neurol Sci 2000; 21(Suppl):S71–S73Crossref, Medline, Google Scholar
25 : Magnetic resonance techniques in neuronal ceroid lipofuscinoses and some other lysosomal diseases affecting the brain. Curr Opin Neurol 1997; 10:519–524Crossref, Medline, Google Scholar
26 : Early differential diagnosis of infantile neuronal ceroid lipofuscinosis, Rett syndrome, and Krabbe disease by CT and MR. AJNR Am J Neuroradiol 1994; 15:1443–1453Medline, Google Scholar
27 : MRI evaluation of the brain in infantile neuronal ceroid-lipofuscinosis, Part 2: MRI findings in 21 patients. J Child Neurol 1995; 10:444–450Crossref, Medline, Google Scholar
28 : Neuronal Ceroid-Lipofuscinoses. GeneReviews. Edited by Pagon RABird TDDolan CR. Seattle, WA, University of Washington, Seattle 1993–2010. [Internet]Google Scholar
29 : Diagnosis of the neuronal ceroid lipofuscinoses: an update. Biochim Biophys Acta 2006; 1762:865–872Crossref, Medline, Google Scholar

