
The Neuropsychiatric Manifestations of Huntington’s Disease-Like 2
Abstract
Huntington’s disease-like 2 (HDL2) is a rare neuropsychiatric disorder that resembles HD but results from a distinct mutation. The authors present a patient with HDL2, hospitalized for psychiatric management, and they review the neuropsychiatric manifestations of this disorder. Depression, irritability/aggression, and frontal lobe personality changes are common presentations of HDL2 and are comparable to classic HD. Patients with HDL2 may differ from those with HD in having a lower incidence of obsessive-compulsive acts, known suicides, antisocial acts, and changes in sexuality. Clinicians should be aware of the psychiatric presentations of this disorder, when to obtain genetic testing, and how to manage problematic behaviors.
Huntington's disease (HD) is an autosomal dominant neurodegenerative disorder, characterized by the classic triad of movement, psychiatric, and cognitive abnormalities.1,2 Huntington's disease-like 2 (HDL2) is also a progressive neurodegenerative disorder, with similar clinical and radiological features as HD,2 including movement, psychiatric, and cognitive abnormalities. HDL2 has a parkinsonian variant similar to the juvenile-onset (Westphal) variant of HD. HDL2 is one of several neurodegenerative disorders with findings phenotypically similar to HD, but without the characteristic HD gene.1 Whereas HD is due to a cytosine-adenine-guanine (CAG) trinucleotide repeat expansion on chromosome 4, the etiology of HDL2 is due to a cytosine-thymine-guanine (CTG)/CAG expansion mutation on chromosome 16q24.3 of the Junctophilin-3 gene (JPH3).1–5
We report a patient with HDL2 who underwent neuropsychological testing and functional neuroimaging, and we review the literature on the neuropsychiatric manifestations of this disorder as compared with HD. We conclude that, compared with HD, the psychiatric and cognitive abnormalities of HDL2 share many features and differ in others. Knowledge of HDL2 can help clinicians in the diagnosis, genetic testing, and management of patients with this disorder.
Case Report
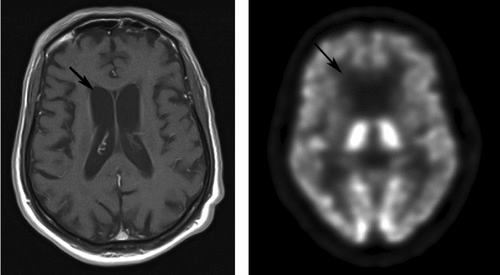
A 63-year-old, right-handed African American man was hospitalized for depression, a personality change, and a movement disorder. His illness began at age 55 years with progressive gait difficulty. At age 60, he sought care for dysphoria and feelings of hopelessness related to his as-yet undiagnosed, movement disorder. His motor symptoms had progressively worsened, resulting in ballistic movements of all limbs, choreoathetosis, and dysphagia, plus a decrease in memory and cognition. In addition to diffuse cortical atrophy, magnetic resonance imaging (MRI) demonstrated barely visible caudate nuclei bilaterally, with thin basal ganglia (Figure 1). Subsequent fluorodeoxy-glucose positron emission tomography (PET) demonstrated markedly decreased metabolic activity bilaterally in the caudate nuclei and putamen (Figure 1).
At age 61, he had been hospitalized with major depression, personality changes, and a further decline in memory and cognition. Before onset of HDL2, others described the patient as having a calm and agreeable nature. He was now despondent, emotionally labile, anxious, irritable, angry, and argumentative, and he showed perseverative behavior. His family history was positive for parkinson-like problems, with shuffling gait and late-life dementia in his mother and “jerking movements” in a male cousin. On examination, he had minimal verbal output, with prominent choreoathetosis. HD was suspected, but genetic testing was negative. Additional genetic testing revealed 15/43 trinucleotide repeats of JPH3 (reference range: <29: Normal; 29–39: Intermediate; >40: Abnormal), consistent with the diagnosis of HDL2.
Over the next 2 years, he manifested increases in agitation; verbal aggression, including yelling; and physical aggression, including kicking in a door. The patient was frequently demanding toward others. At one point, he held a butter knife to his neck and threatened suicide if his needs were not met. He was started on haloperidol, with mild improvement in motor function but minimal effect on his behavior.
Progressive worsening of his behavior had prompted the current hospitalization at age 63. On admission, he had flat affect, mild bradykinesia, orobucchal dyskinesia, moderate general choreoathetosis, intermittent dystonia in trunk and upper limbs, hand tremors, and unsteady gait. Neuropsychological mental status examination revealed multiple deficits (Table 1). He had intact basic attention but impaired working memory. Language was nonfluent, with sparse, hypophonic, dysarthric, and aprosodic verbal output, but without paraphasic errors. His comprehension was impaired for passive possessives and for reading. Episodic, declarative memory was impaired, but he had moderately preserved remote memory for significant historical events. Praxis was intact, but the patient failed at calculations and the clock-drawing task. Finally, on frontal-executive tasks, he had difficulties with similarities, idioms, and proverbs, and with alternating programs, alternate tapping, and the Go/No-Go test.
| Patient at Age 63 | |
|---|---|
| Mini-Mental State Exam (max: 30) | 17 |
| Montreal Cognitive Assessment (max: 30) | 9 |
| Digit-Span Forward (Normal: ≥5) | 5 |
| Digit-Span Reverse | 3 |
| Months-in-Reverse | 0 |
| “A” Vigilance Test | 0 omissions or comissions |
| Serial “3’s” Test | “15, 6, 0” |
| Verbal Fluency | |
| Animals/minute (Normal: ≥12) | 6 |
| “F” words/minute (Normal: ≥10) | 4 |
| Mini-Boston Naming Test15 | 11 |
| Auditory Verbal Learning Test | 0 |
| Delayed Recall (Normal: ≥7/10) | 10 (5 False Positives) |
| Recognition (max: 20; Normal: ≥16/20) | |
| Visuospatial8 | 4 |
| Frontal Assessment Battery (max: 18) | 9 |
| Idioms and Proverbs3 | 0 |
The patient was treated with low-dose citalopram and gabapentin, as well as continuation of the haloperidol. Over the next few months, his emotional lability, anxiety, irritability, argumentativeness, and verbal and physical aggression subsided, with only occasional instances of agitation. There was modest improvement in his cognitive test results as his behavior improved.
Discussion
In this patient, the initial neuropsychiatric symptoms were dysphoria, hopelessness, and eventual major depression. As his HDL2 progressed, he also developed a personality change, with irritability, argumentativeness, perseverative behavior, and significant aggression. In addition to psychiatric symptoms, the patient had cognitive deficits in mental control, verbal fluency, memory retrieval, visuospatial constructions, calculation, and several frontal systems tasks.
Recognizing patients like this one with HDL2 requires a high index of suspicion. In the United States, HDL2 comprises only about 1% of cases referred for HD testing who prove to be negative for HD.6 HDL2 is most common among black South Africans and occurs almost exclusively in individuals with African ancestry.4 There are cases, however, with apparent European ancestry, although bearing an African HDL2 haplotype,7 and HDL2 occasionally occurs among North American and Japanese patients with HD-like clinical presentations.4
There are two variants of HDL2, characterized by differences in movement abnormalities. The first variant has prominent rigidity and parkinsonism and is comparatively similar to juvenile-onset HD, whereas the second, more common variant, has prominent choreoathetosis and closely resembles adult-onset HD.2,6 The common variant of HDL2 usually presents between age 29 and 41 with gait or coordination difficulty and progresses to severe dementia and death in 10–15 years.6 This patient’s later age at onset probably relates to his relatively low number of nucleotide repeats (43; Abnormal >40); as in HD, the more nucleotide repeats, the earlier the age at onset.
HDL2 is only one of several neurodegenerative disorders that are phenotypically similar to HD. HDL1 is an autosomal dominant neurodegenerative familial prion disease.2,8 The disorder is caused by 168 or 192 base-pair insertions, encoding extra octapeptide repeat insertions in the PRNP gene, resulting in a clinical picture of abnormal movements, dementia, and personality and psychiatric abnormalities similar to those of HD.8 There is an HDL3 disorder, which is an autosomal recessive HD-like disorder with a poorly understood genetic basis. There is also an HDL4 (spinocerebellar ataxia 12 or SCA12), which is attributable to mutation in the gene encoding the TATA box-binding protein (TBP).2,8 Furthermore, SCA1, SCA2, dentatorubral-pallidolyusian atrophy, choreoacanthocytosis, and neurodegeneration with brain iron accumulation are all disorders that resemble HD, but lack the characteristic CAG expansion.2,8
Psychiatric symptoms appear to be universal in HDL2 (see Table 2).1,4 Depression, anxiety, irritability and agitation, perseverative behavior, delusions and hallucinations, aggressive behavior, and frontal disinhibition or apathy have occurred in patients with HDL2.1,3,9,10 In the largest reported series of HDL2 subjects, psychiatric symptoms occurred in 100% of the affected individuals; these included depression, anxiety, irritability, perseverative behavior, delusions, and, possibly, hallucinations.1 In another series of HDL2 patients from Brazil, the investigators reported depression, aggressive behavior, visual hallucinations, and social withdrawal.3 Additional small series or case reports describe prominent depression, frontal behavioral changes, delusions, and paranoia in patients with this disorder.9–11
| Neuropsychiatric Symptoms | HDL2a | HDb |
|---|---|---|
| Cognitive decline/dementia | 90%+ | 100% |
| Irritability/agitation/aggression | 15%–37%+ | 38%–73% |
| Mood disorders (with/without anxiety) | 56%+ | 33%–69% |
| Frontal lobe personality changes (apathy and/or disinhibition) | 11%–37%+ | 34%–76% |
| Psychosis (delusions and/or hallucinations) | 37%+ | 3%–12% |
| Obsessive-compulsive behavior | No datac | 10%–52% |
| Sexuality (hyper-, hypo-, paraphilias) | No data | 2.5%–29.4% |
| Suicide | No data | 6%–8% of deathsd |
| Mania | No data | 2.0%–12.5% |
Given the similarities of HDL2 and HD, a comparison of their psychiatric and cognitive symptoms may serve to better explain HDL2 (see Table 2). Although they both have cortical atrophy, striatal neuronal loss and gliosis, amygdalar involvement, and intranuclear protein aggregates, the distribution of these aggregates is not identical and may underlie differences in neuropsychiatric manifestations between HDL2 and HD.6 Like those with HLD2, patients with HD present with depression, irritability, agitation, or social withdrawal.12 Depression and irritability appear to occur before motor symptoms in many HD patients,12 and subtle, subclinical psychiatric symptoms occur in prediagnosed individuals with the HD mutation up to 10 years before their diagnosis.13 Aggression and apathy are additional common features in both HD and HDL2. Similar to HD, in HDL2, psychiatric symptoms such as aggressiveness can be most prominent at the early stages, whereas the later stages can be more characterized by apathy. Obsessive-compulsive features may be more common in HD than in HDL2, but the data on HDL2 are scarce, and many HDL2 patients have prominent perseverations.14 Also, investigators have not, as yet, reported comparably high suicide rates, frequency of antisocial acts, and changes in sexuality in HDL2, as compared with HD.14,15
In terms of treatment, this patient's behavioral symptoms responded to citalopram, a selective serotonin reuptake inhibitor. Other investigators have found that depression among patients with HDL2 can respond in part to sertraline and nortriptyline.10 In HD, additional success has been found with bupropion, venlafaxine, and nefazodone, as well as antipsychotics for agitation, hallucinations, and delusions,12 and this may be the case for HDL2, as well. Unfortunately, this patient had only a partial response to haloperidol.
In conclusion, this patient adds to the small but growing literature on the neuropsychiatry of HDL2. Although similar to classic HD, this disorder may prove to have differences in certain clinical manifestations and, possibly, in management. Much more research needs to be focused on this eminently neuropsychiatric disorder in order to clarify these and other aspects of HDL2.
1 : A disorder similar to Huntington's disease is associated with a novel CAG repeat expansion. Ann Neurol 2001; 50:373–380Crossref, Google Scholar
2 : Huntington’s disease phenocopy syndromes. Curr Opin Neurol 2007; 20:681–687Crossref, Medline, Google Scholar
3 : Clinical and genetic analysis of 29 Brazilian patients with Huntington’s disease-like phenotype. Arq Neuropsiquiatr 2011; 69:419–423Crossref, Medline, Google Scholar
4 : Huntington’s Disease-like 2 (HDL2) in North America and Japan. Ann Neurol 2004; 56:670–674Crossref, Medline, Google Scholar
5 : A repeat expansion in the gene encoding junctophilin-3 is associated with Huntington disease-like 2. Nat Genet 2001; 29:377–378Crossref, Medline, Google Scholar
6 : A comparison of Huntington’s disease and Huntington’s disease-like 2 neuropathology. J Neuropathol Exp Neurol 2008; 67:366–374Crossref, Medline, Google Scholar
7 : Huntington disease-like 2: the first patient with apparent European ancestry. Clin Genet 2008; 73:480–485Crossref, Medline, Google Scholar
8 : The Huntington’s disease-like syndromes: what to consider in patients with a negative Huntington’s disease gene test. Nat Clin Pract Neurol 2007; 3:517–525Crossref, Medline, Google Scholar
9 : Huntington’s disease-like phenotype due to trinucleotide repeat expansions in the TBP and JPH3 genes. Brain 2003; 126:1599–1603Crossref, Medline, Google Scholar
10 : Phenotypic features of Huntington’s disease-like 2. Mov Disord 2003; 18:1527–1530Crossref, Medline, Google Scholar
11 : A South African mixed-ancestry family with Huntington disease-like 2: clinical and genetic features. Mov Disord 2007; 22:2083–2089Crossref, Medline, Google Scholar
12 : Neuropsychiatry of Huntington’s disease and other basal ganglia disorders. Psychosomatics 2000; 41:24–30Crossref, Medline, Google Scholar
13 : Psychiatric symptoms in Huntington’s disease before diagnosis: The Predict-HD Study. Biol Psychiatry 2007; 62:1341–1346Crossref, Medline, Google Scholar
14 : Psychopathology in verified Huntington's disease gene carriers. J Neuropsychiatry Clin Neurosci 2007; 19:441–448Crossref, Medline, Google Scholar
15 : Huntington’s disease: update and review of neuropsychiatric aspects. Int J Psychiatry Med 1994; 24:189–208Crossref, Medline, Google Scholar

