
Klüver-Bucy Syndrome After Bilateral Selective Damage of Amygdala and Its Cortical Connections
Abstract
Isolated symmetric damage to the amygdalae and their cortical connections occurred in an individual following cancer treatment. The lesions were imaged after reversal of hyponatremia. The patient displayed marked behavioral changes including visual agnosia, hypersexuality, hyperorality, a tendency to react to every visual stimulus, and memory deficits. The cluster of neurobehavioral symptoms is similar to previously reported accounts of Klüver-Bucy syndrome and suggests the importance of bilateral amygdala involvement in these behavioral changes.
In the 1930s Klüver and Bucy gave their names to an intriguing cluster of behavioral changes that was noted after bilateral removal of the anterior temporal lobes in primates.1 The Klüver-Bucy presentation includes 1) “psychic blindness,” or the inability to recognize the emotional significance of objects; 2) hypersexuality, often directed indiscriminately; 3) altered emotional behavior, particularly placidity; 4) hyperorality and the ingestion of inappropriate objects (pica); 5) “hypermetamorphosis,” or the tendency to react to every visual stimulus; and 6) memory deficits. Other researchers subsequently associated many of these behavioral changes to bilateral amygdala ablation,2 although one report exists of this syndrome occurring following unilateral anterior temporal resection.3
Full-blown Klüver-Bucy syndrome (KBS) in humans is rare, and usually extensive lesions and a generalized dementia are present. The first published identification of a human patient with KBS was made in 1955 by Terzian and Dalle Ore4 in an epileptic who underwent bilateral temporal lobectomy. Partial syndromes are often reported, and the published criteria for this diagnosis vary. Many authors have pointed out the differences in the presentation of symptoms between humans and nonhuman primates. Lilly et al.5 attribute the more complex behavioral syndrome seen in humans to the evolutionary advances of the human brain: psychic blindness becomes prosopagnosia or other forms of visual agnosia; hyperorality becomes hyperphagia and bulimia; and hypermetamorphosis becomes distractibility. In addition, KBS in humans is inevitably associated with aphasia (most commonly a receptive Wernicke's aphasia) and amnesia.5
Alterations in emotional behavior in humans include apathy, lethargy, and emotional unresponsiveness. Hypersexuality is one of the least frequently reported symptoms in humans.
Copulation, masturbation, and other self-stimulation all are uncommon, although inappropriate sexual remarks and gestures are frequent.6 Changes in sexual preference have also been reported.5 By convention, KBS is generally diagnosed if at least three of the characteristic symptoms are present.7 The most commonly reported symptoms are hyperorality and placidity; hypersexuality and the sensory agnosias are least frequently reported.5 The most common etiology of KBS in humans is herpes encephalitis, although trauma, hypoxia, and a wide variety of central nervous system diseases that can affect medial temporal lobe structures, such as Pick's disease, have been reported.8
Various theories have been suggested to explain this syndrome. Poeck9 advanced an ontologic and evolutionary explanation of KBS, and Geschwind10 viewed KBS as a disconnection syndrome produced by interrupting visual input to the limbic system. Bear11 attributed KBS symptomatology to an imbalance between complementary dorsal and ventral cortical visuolimbic systems. Lesions in the ventral (temporofrontal) system can disrupt access to learned emotional associations and responses, including inhibitory social restraints. This lack of access to learned responses leaves the dorsal (parietofrontal) system, which directs surveillance of the environment for drive-relevant stimuli, unopposed, causing hyperactive nonspecific exploration. It is known that bilateral lesions in the amygdala by themselves will produce the visual agnosia component of KBS in animals,12 and disorders of facial recognition in humans have been seen following bilateral amygdalotomy.13 Lesions in both the inferior temporal cortex and the superior temporal polysensory area mimic the deficits observed after amygdala ablation, implying that these two regions supply the amygdala with information that identifies objects as being edible or aversive.14 LeDoux15 and others have elucidated a more precise role of the amygdala in fear conditioning and in the learning of emotional significance of stimuli through stimulus-reward associations. Recent psychopharmacologic studies in primates suggest that lesions of the temporal pole disconnect excitatory projections to the amygdala, causing reduced dopamine and serotonin levels and increased norepinephrine levels in the amygdala.16
The authors have encountered a patient, described below, who experienced bilateral damage to the amygdala and both insular and temporal gyri with relative sparing of adjacent limbic structures. This unusual pattern of damage suggested selective neurotoxicity primarily in the basolateral amygdala and its cortical connections. Behavioral symptoms manifested by the patient included hyperorality, visual agnosia, altered emotional status, and Wernicke's aphasia.
CASE REPORT
A.B. was a 43-year-old white man. He was married and had a 4-year-old son. A.B. had attended college for 2 years and was employed as an operator at a water plant. His past medical and psychiatric histories were unremarkable.
A.B. originally consulted his physician in early January 199 with complaints of sharp pain in his upper chest. He also complained of progressive weakness, sore throat, anorexia, and mild fevers. An ultrasound scan of his gall bladder revealed an enlarged liver and spleen. A liver biopsy was performed, and in February a diagnosis of metastatic malignant melanoma was made. Prior to the pathological confirmation of his disease, A.B. began intra-arterial therapy for presumed hepatocellular carcinoma. This treatment included 5-fluorouracil, doxorubicin, and cisplatin. Following the diagnosis of melanoma, this treatment was discontinued, and he was treated with dacarbazine and interferon alpha. He was subsequently treated with two more courses of chemotherapy as an outpatient. A.B.'s response to this chemotherapy was marked by a reduction in liver size and improvement of his symptoms until early April, when he noted a rapid return of symptoms. A.B. was admitted to the hospital for another course of intra-arterial chemotherapy with doxorubicin, cisplatin, and dacarbazine in mid-April.
On April 22 A.B. was taken to the emergency room and was admitted to the hospital after experiencing a generalized seizure. Laboratory test results revealed a low sodium level (102 mEq/l) and pancytopenia (white blood cell count of 700/mm3, hemoglobin level of 8.4 gm/dl, and platelet count of 58,000/mm3). Slightly elevated protein levels (74 mg/dl) in the spinal fluid of the patient were also noted. A.B. was treated with granulocyte colony–stimulating factor (G-CSF), imipenem, and phenytoin. The hyponatremia was corrected over 48 hours to 134 mEq/l. Other medications included corticosteroids, lorazepam (for sedation), and chloral hydrate (for sleep). The patient was also given acyclovir as an antiviral agent pending the results of additional spinal fluid studies.
The patient's mental status and orientation gradually improved from the time of his admission until the afternoon of April 24, at which time he became progressively more confused and lethargic. His mental status remained impaired throughout the remainder of his hospitalization. For this condition, he was treated with haloperidol for 2 days (10 mg q 6 hrs) and was then switched to chlorpromazine (50 mg im). During this time (late April and early May), A.B.'s speech did not usually make sense, and many neologisms were present. He uncharacteristically used obscene language and did not recognize his mother, wife, or son. He could not follow commands and did not respond appropriately to questions or other input. He often put objects in his mouth and was observed eating polystyrene containers. On occasion, his behavior was inappropriate and violent: spitting at his son, using foul language, biting people, and trying to kiss his nurse. He sometimes talked of going to a place that he had visited as a child, and he would occasionally pick up the phone and talk as if someone were on the line. His mental state did not appear to be due to a delirium in that it was not transient and was not associated with disturbed arousal, disruption of his sleep–wake cycle, psychomotor agitation, or outright illusions or hallucinations.17
MRI scan performed on April 29 was normal. EEG obtained on the same day revealed marked slowing of waves over the entire brain and especially in the frontal regions. However, another MRI scan 9 days later revealed abnormal signal in the amygdalae, hippocampi, and posterior parahippocampal gyri. Damage to the blood–brain barrier, evidenced by gadolinium enhancement, was seen in the amygdala, insular, and temporal gyri, with relative sparing of the parahippocampus and hippocampus structures (Figure 1). Analysis of spinal fluid on April 30 was unremarkable. A brain biopsy specimen was not obtained.
The patient was transferred to an inpatient hospice unit on May 13. At the time of his discharge from the hospital, A.B.'s laboratory values had improved (white blood cell count, 11,900/mm3; platelet count, 279,000/mm3; hemoglobin, 11.5 gm/dl).
On May 20, a behavioral assessment was attempted. However, A.B. had received lorazepam 2 hours previously and was difficult to arouse. He was unresponsive to commands and appeared unable to comprehend speech. When asked to squeeze the examiner's hand, he attempted to kiss it. His responses to questions were fluent but made no sense. His spontaneous writing was also fluent but contained word substitutions, perseverations, and nonsense words. A second behavioral examination was conducted on the following day with the lorazepam withheld. A.B. was more alert and talkative and was found to be feeding himself appropriately. His Wernicke's aphasia persisted; he could not follow one-step commands or name even simple objects. When he was given a comb, he called it a “saw,” smelled it, wiped it off with his napkin, and set it down. When he was given an apple and a ball, he smelled them, set them down, and said that they smelled good. A.B. did not appear to recognize a cup when it was handed to him, although he was observed to drink coffee appropriately from another cup. When he was asked to write his name, he wrote a phone number (with extra numbers). When he was asked to copy a square, he drew one contiguous with the model and perseverated several other squares. A.B. did not repeat words and did not respond to a ringing phone. He shook the examiner's hand when she offered it at the end of the interview.
A.B.'s fluent nonsensical speech, word substitutions, and lack of comprehension indicated a Wernicke's-type aphasia. A.B. could not read, write, or repeat words during the examination. The neurobehavioral evaluation suggested that A.B. was experiencing visual agnosia, meaning that he did not recognize objects (such as the comb and cup) and therefore did not use them appropriately. Although he was observed to be mildly apraxic, he did intermittently demonstrate adequate motor sequencing for object usage, such as picking up the phone and shaking the examiner's hand, suggesting that apraxia could not fully account for his inability to visually recognize and distinguish familiar objects. He had a tendency to examine objects orally; during the first assessment he put everything in his mouth and had been observed eating nonfood objects. During the second assessment he smelled all the objects he was given. According to caregiver reports, his conduct varied between placidity and inappropriately violent or sexual behaviors. This constellation of symptoms (visual agnosia, Wernicke's aphasia, oral exploratory behavior, and altered emotional status), combined with MRI scan results that revealed bilateral mesiotemporal lobe lesions, was consistent with Klüver-Bucy syndrome. Although anecdotal reports of A.B.'s behavior and the presence of bilateral temporal lesions suggest the likelihood of memory deficits, the severity of his language and visual disturbances precluded a formal assessment of memory. Similarly, in light of A.B.'s other cognitive impairments, it was difficult to establish the presence of hypermetamorphosis.
Over the next 3 months A.B.'s condition continued to worsen, and he died quietly in August.
DISCUSSION
This report describes a patient with a unique constellation of limbic abnormalities that resulted in a Klüver-Bucy–like syndrome. The etiology of this neurologic injury is not clear. Most of the patient's chemotherapy was delivered by intra-arterial infusion, which is thought to reduce systemic toxicity of the drugs.18 The neurotoxic effects of systemic interferon alpha are well described19 and occur in more than half of patients receiving multi-agent chemotherapy with interferon for metastatic melanoma.20 However, there are no reports of severe focal neurologic effects such as are reported here. It is known that individuals with preexisting dihydropyrimidine dehydrogenase deficiency can develop severe encephalopathy with demyelination from 5-fluorouracil,21 which is a possibility in this case. Leptomeningeal spread of cancer is not likely because the patient did not have malignant cells in his cerebrospinal fluid and the lesions were not seen on the initial MRI scan. Infection is also not likely because there were no white blood cells present in the spinal fluid, and the patient was placed on prophylactic acyclovir and did not have a fever or other signs of infection. Another possibility, paraneoplastic limbic encephalopathy, is commonly associated with small cell lung cancer22 but has not been described with melanoma. The paraneoplastic syndrome primarily associated with melanoma is retinopathy.23 The most likely explanation for this individual's neurologic injury is extrapontine myelinolysis secondary to rapid correction of hyponatremia (32 mEq/l over 48 hours). Although the majority of patients with this disorder develop symmetric pontine lesions, bilateral thalamic and cortical lesions have also been described, and the development of the lesions 9 days after the onset of the clinical presentation is typical.24
The lesions in this case selectively involved the basolateral amygdala and its connections to the cortical neurons in the insula and the temporal gyri, with less severe involvement of the hippocampus and parahippocampal areas. There was sparing of the extended amygdala (centromedial amygdaloid nucleus, stria terminalis) and striatal connections. This is possibly because the basolateral part of the amygdala is not laminated and its histology, histochemistry, and connections are in many ways reminiscent of a cortical structure. It is distinctly different from the centromedial part of the amygdaloid body and the extended amygdaloid complex.
It has been assumed that extensive lesions are required to cause Klüver-Bucy syndrome in humans, including lesions not only of the amygdala and uncus, but also the hippocampus, the cingulate gyrus, and the orbitofrontal, insular, and temporal cortices.25 This case differs from other reports of KBS in humans in that the anatomic abnormalities revealed by MRI were selective and the syndrome did not occur in the setting of global dementia,26–28 large areas of infarction,7,29 herniation from hemorrhage,6 or extensive limbic degeneration seen in amyotrophic lateral sclerosis.30 There are no other reports of the development of KBS or bilateral amygdala lesions caused by post-hyponatremia myelinolysis. Moreover, this report adds additional information to our conceptualization of the relationship between limbic structures and human behavior.
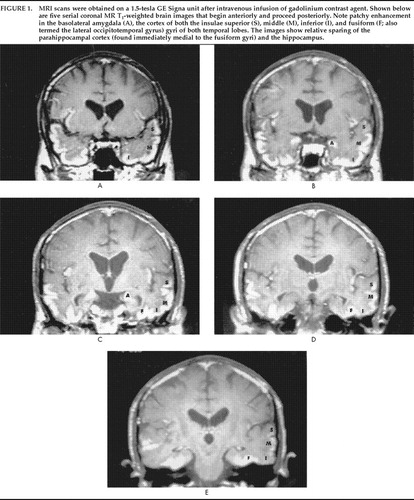
FIGURE 1. MRI scans were obtained on a 1.5-tesla GE Signa unit after intravenous infusion of gadolinium contrast agent. Shown below are five serial coronal MR T1-weighted brain images that begin anteriorly and proceed posteriorly. Note patchy enhancement in the basolateral amygdala (A), the cortex of both the insulae superior (S), middle (M), inferior (I), and fusiform (F; also termed the lateral occipitotemporal gyrus) gyri of both temporal lobes. The images show relative sparing of the parahippocampal cortex (found immediately medial to the fusiform gyri) and the hippocampus.
1. Klüver H, Bucy PC: Preliminary analysis of functions of the temporal lobes in monkeys. Arch Neurol Psychiatry 1939; 42:979–1000Crossref, Google Scholar
2. Hart RP, Kwentus JA, Frazier RB, et al: Natural history of Klüver-Bucy syndrome after treated herpes encephalitis. South Med J 1986; 79:1376–1378Crossref, Medline, Google Scholar
3. Ghika-Schmid F, Assal G, DeTribolet N, et al: Klüver-Bucy syndrome after left anterior temporal resection. Neuropsychologia 1995; 33:101–113Crossref, Medline, Google Scholar
4. Terzian H, Dalle Ore G: Syndrome of Klüver and Bucy reproduced in man by bilateral removal of the temporal lobes. Neurology 1955; 5:373–380Crossref, Medline, Google Scholar
5. Lilly R, Cummings JL, Benson F, et al: The human Klüver-Bucy syndrome. Neurology 1983; 33:1141–1145Crossref, Medline, Google Scholar
6. Rossitch E, Carrazana EJ, Ellenbogen R, et al: Klüver-Bucy syndrome following recovery from transtentorial herniation. Br J Neurosurg 1989; 3:503–506Crossref, Medline, Google Scholar
7. Oliveira V, Ferro JM, Foreid JP, et al: Klüver-Bucy syndrome in systemic lupus erythematosus. J Neurol 1989; 236:55–56Crossref, Medline, Google Scholar
8. Conlon P, Kertesz A, Mount J: Klüver-Bucy syndrome with severe amnesia secondary to herpes encephalitis. Can J Psychiatry 1988; 33:754–756Crossref, Medline, Google Scholar
9. Poeck K: Klüver-Bucy syndrome, in The Handbook of Clinical Neurology, edited by Venken PJ, Bruin GW. New York, Elsevier, 1969, pp 343–367Google Scholar
10. Geschwind N: Disconnection syndromes in animals and man. Brain 1965; 88:237–294Crossref, Medline, Google Scholar
11. Bear DM: Hemispheric specialization and the neurology of emotion. Arch Neurol 1983; 40:195–202Crossref, Medline, Google Scholar
12. Zola-Morgan S, Squire LR, Amaral DG: Lesions of the amygdala that spare adjacent cortical regions do not impair memory or exacerbate the impairment following lesions of the hippocampal formation. J Neurosci 1989; 9:1922–1936Crossref, Medline, Google Scholar
13. Jacobson R: Disorders of facial recognition, social behaviour and affect after combined bilateral amygdalotomy and subcaudate tractotomy: a clinical and experimental study. Psychol Med 1986; 16:439–450Crossref, Medline, Google Scholar
14. Aggleton JP, Mishkin M: Visual impairments in macaques following inferior temporal lesions are exacerbated selectively by additional damage to superior temporal sulcus. Behav Brain Res 1990; 39:262–274Crossref, Medline, Google Scholar
15. LeDoux JE: Brain mechanisms of emotion and emotional learning. Curr Opin Neurobiol 1992; 2:191–197Crossref, Medline, Google Scholar
16. Kling AS, Tachiki K, Lloyd R: Neurochemical correlates of the Klüver-Bucy syndrome by in vivo microdialysis in monkey. Behav Brain Res 1993; 30:161–170Crossref, Google Scholar
17. Olofsson SM, Weitzner MA, Valentine AD, et al: A retrospective study of the psychiatric management and outcome of delirium in the cancer patient. Support Care Cancer 1996; 4:351–357Crossref, Medline, Google Scholar
18. Vexler AM, Mou X, Gabizon AA, et al: Reduction of the systemic toxicity of cisplatin by intra-arterial hepatic route administration for liver malignancies. Int J Cancer 1995; 60:611–615Crossref, Medline, Google Scholar
19. Pavol MA, Meyers CA, Rexer JL, et al: Pattern of neurobehavioral deficits associated with interferon-alpha therapy for leukemia. Neurology 1995; 45:947–950Crossref, Medline, Google Scholar
20. Vuoristo M-S, Gröhn P, Kellokumpu-Lehtinen P, et al: Intermittent interferon and polychemotherapy in metastatic melanoma. J Cancer Res Clin Oncol 1995; 121:175–180Crossref, Medline, Google Scholar
21. Morrison GB, Bastian A, Dela Rosa T, et al: Dihydropyrimidine dehydrogenase deficiency: a pharmacogenetic defect causing severe adverse reactions to 5-fluorouracil-based chemotherapy. Oncology Nursing Forum 1997; 24:83–88Medline, Google Scholar
22. Patel AM, Davil DG, Peters SG: Paraneoplastic syndromes associated with lung cancer. Mayo Clin Proc 1993; 68:278–287Crossref, Medline, Google Scholar
23. Ohguro H, Takaya T, Ogawa K, et al: Cancer-associated retinopathy. Nippon Ganka Gakkai Zasshi–Acta Societatis 1997; 101:283–287Medline, Google Scholar
24. Laureno R, Karp BI: Myelinolysis after correction of hyponatremia. Ann Intern Med 1997; 126:57–62Crossref, Medline, Google Scholar
25. Halgren E: Emotional neurophysiology of the amygdala within the context of human cognition, in The Amygdala, edited by Aggleton JP. New York, Wiley-Liss, 1992, pp 191–228Google Scholar
26. Lanska DJ, Cunier RD, Cohen M, et al: Familial progressive subcortical gliosis. Neurology 1994; 44:1633–1643Crossref, Medline, Google Scholar
27. Poeck K: The Klüver-Bucy syndrome in man, in The Handbook of Clinical Neurology, edited by Frederiks JAM. New York, Elsevier, 1985, pp 257–263Google Scholar
28. Forstl H, Burns A, Levy R, et al: Neuropathological correlates of behavioural disturbance in confirmed Alzheimer's disease. Br J Psychiatry 1993; 163:364–368Crossref, Medline, Google Scholar
29. Fragassi NA, Longobardi T, Pellegrino MG, et al: Klüver-Bucy syndrome: a case report. Acta Neurologica (Napoli) 1990; 12:138–142Medline, Google Scholar
30. Dickson DW, Horoupian DS, Thal LJ, et al: Klüver-Bucy syndrome and amyotrophic lateral sclerosis: a case report with biochemistry, morphometrics, and Golgi study. Neurology 1986; 36:1323–1329Crossref, Medline, Google Scholar

