
Agrypnia Excitata: Current Concepts and Future Prospects in Management
“
Fatal Familial Insomnia
Fatal familial insomnia (FFI) is an extremely rare neurodegenerative prion disease. 3 It is transmitted in an autosomal dominant pattern and is characterized by behavioral problems, autonomic instability, hallucinations, and ultimately death. 3 The disease arises from a mutation in the gene responsible for the expression of a protein resulting in the generation of insoluble proteins called prion proteins that contribute to the deterioration of vital neurological structures within the CNS. 2 Over time these insoluble proteins congregate to form amyloid plaques throughout the CNS, particularly in the areas within or near the hypothalamus, leading to the characteristic deficits in hypothalamic function in individuals afflicted with FFI. Fatal familial insomnia is characterized by a devastating clinical course that includes recurrent sleep disorders, neurological dysfunction, and eventual death. Because of the extreme rarity of FFI, a paucity of reliable data exists on approaches and maximal treatment strategies for this disorder. The following is a thorough examination of FFI with a particular focus placed on current concepts in diagnosis and management as well as future prospects for the condition.
Clinical Manifestations
Fatal familial insomnia is characterized by behavioral disturbances, ataxia, pyramidal signs, autonomic problems, and widespread neurological compromise. 3 After the initial onset of symptoms, FFI evolves rapidly, and death occurs months after the first symptom sets in. 3 , 4 The symptoms of FFI are directly related to the area of neurological involvement in the disorder. Since the thalamus and associated thalamic structures are the most affected in FFI, it stands to reason that bodily functions controlled by the thalamus are commensurately affected in patients with FFI. The thalamus is a prominent mass of gray matter located above the brainstem that controls bodily functions including motor control and somatosensory and visual sensory signals. It also relays sensory signals to and from the cerebral cortex and vital anatomical structures throughout the body. Typically, patients complain of persistent insomnia, frequent nocturnal awakenings, and persistent daytime fatigue. 1 , 5 , 6 Autonomic activation in the form of excessive salivation and perspiration along with elevated blood pressure, tachycardia, pyrexia, and appetite disturbances are also common. 1 , 6 , 7 Bizarre dreams often occur and tend to be superseded by a stupor-like state that eventually leads to coma and death. 1
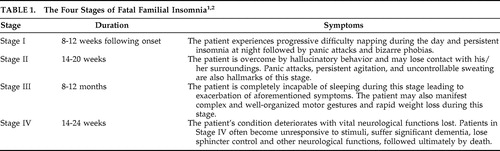
During sleep, the ability of the thalamus to effectively relay such signals becomes compromised, allowing the body to succumb to the sleep state. Therefore, the thalamic dysfunction that ensues in FFI yields a myriad of problems associated with the aforementioned thalamic functions, namely those related to or affected by sleep. The disease runs a uniformly fatal course that manifests in four stages ( Table 1 ). Ultimately, patients expire 18 months after the onset of symptoms. 2 Common clinical features of FFI include hallucinations, phobias, dementia, decreased reflexes, and an inability to lacrimate or appreciate pain. 1 , 2 Later stages of the disease lead to autonomic dysfunction, which manifests as dementia, coma, and loss of sphincter control. 1 The wide array of symptoms in FFI has led experts to classify it as a pleiotropic disease, or a disease with one mutant gene that yields one mutant protein but results in many phenotypic abnormalities. The pleiotropic basis of the disorder may account for the significant variation in disease presentation from individual to individual, even in patients from the same family.
Epidemiology
Fatal familial insomnia is transmitted via autosomal dominant inheritance, meaning it affects both genders with equal frequency. 8 , 9 On average, individuals with the mutant gene most often develop symptoms between the ages of 40 to 60 years, with an average of onset at 51 years old. 10 Death ensues by 32–72 weeks after first onset of symptoms. 5 – 7 , 10
Pathophysiology
Fatal familial insomnia results from a genetic mutation in the prion protein gene, or PRNP gene, that results in the replacement of asparagine with aspartic acid at codon 178, along with substitution of asparagine for methionine at codon 129. 1 , 3 , 11 Although the exact function of the PRNP gene remains uncertain, it is likely involved in the cell signaling in the CNS. The inborn genetic error in FFI results in a dramatic accumulation of protease resistant prion protein, which is responsible for the cognitive dysfunction that characterizes the disorder. 1 The mutation yields significant thalamolimbic impairment, which is believed to cause the striking phenotypic and clinical features of FFI. 1 , 3 Pathology and imaging studies corroborate this finding by demonstrating widespread hypometabolism throughout the thalami of patients with FFI. 3 Furthermore, the presence of atrophy, gliosis, and neuronal loss in the thalami along with spongiosis in the cerebellum is a common finding in patients with FFI. 1 , 3 , 9 The atrophic changes within the thalamus that characterize FFI are believed to underlie the sleep, autonomic, and circadian disturbances that appear in FFI. 1 , 4
Diagnosis
A number of tests are used to diagnosis FFI. Since FFI is familial in origin, a thorough family history may help in arriving at an accurate diagnosis. A thorough physical examination and detailed history are recommended to rule out systemic or exogenous conditions as a cause of clinical presentation. The physical exam of FFI patients often demonstrates bilateral hyperreflexia and reflex myoclonia. 3 Laryngeal examination may also demonstrate paralysis of the laryngeal cords. 3 Although exact cause of death in FFI remains unclear, experts have pointed to the paralysis of laryngeal cords in FFI as a possible cause of laryngospasm-induced death in such patients. 3 , 4 Given the many neuropsychological sequelae of FFI, comprehensive neuropsychological testing is warranted. Depending on the stage of disease development, the patient may demonstrate any of a wide array of symptoms from hallucinations, behavioral disturbances, autonomic disturbances, alteration in attention and immediate memory, or a propensity to confabulate. 3 The patient’s history will reveal significant sleep disturbances in the form of insomnia and repeated nocturnal awakenings. As with other patients with deterioration of sleep function, a polysomnogram is considered necessary for the diagnosis of FFI and typically reveals the complete absence of spindles and slow-wave sleep along with extended REM periods without muscle atonia. 1 , 3 , 4 , 12 Stridor, nocturnal groaning, loud snoring, and persistent talking and moving may also be present during polysomnogram testing in FFI patients. 3 Finally, given the genetic nature of the disease, genetic testing for mutations in the D178N gene in codon 129 can help confirm the diagnosis. 3 , 8 , 11
Treatment
Although FFI runs a fatal course, recent data suggest that the use of vitamins, sedative-hypnotics (e.g., ethchlorvynol, zolpidem, or diazepam), stimulants, sensory deprivation, exercise, light entrainment, and growth hormone may offer promising methods of managing FFI patients. 13 – 17 Electroconvulsive therapy may improve symptoms in such patients through forced sleep and the use of vitamins, sedative-hypnotics, and stimulants may yield improved quality of life by minimizing stupor-like states that often accompany periods of prolonged sleeplessness. 13 , 14 , 17 Despite improving a patient’s ability to sleep, ECT should be used with caution due to the myriad of side effects such therapy courts, including memory loss and cognitive damage. 13 , 18 , 19 Despite advances in therapeutic strategies and the resultant improvement in symptoms of FFI, a cure for FFI continues to elude clinicians. The need for broader research on the topic is warranted considering the fact that all patients expire within 32–72 weeks after first onset of symptoms. 5 , 7
Morvan’s Chorea
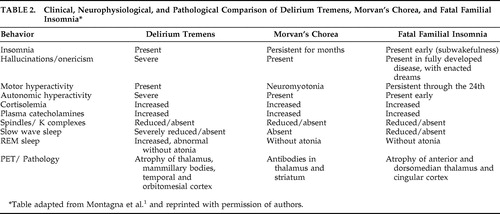
Morvan’s chorea, or “fibrillary chorea,” describes the rare condition in which neuromyotonia (involuntary contraction of muscles at rest) occurs in the setting of peripheral nervous system hyperexcitibility. 1 , 3 , 20 The typical movements in Morvan’s chorea most commonly affect bilateral calves and the posterior part of the thigh. The peripheral nerve hyperexcitibility that dominates in Morvan’s chorea typically results in neuromyotonia as well as hyperhydrosis, paresthesias, dysautonomia, myalgias, incontinence, palpitations, and sleep dysfunction. 3 , 20 , 21 Similar to the other two components of agrypnia excitata, Morvan’s chorea also shares the same clinical features as both FFI and delirium tremens: agrypnia, mental confusion, and a significant reduction of slow wave sleep ( Table 2 ). 1 – 3 , 18 Unlike the other components of agrypnia excitata, however, Morvan’s chorea can arise at any age and may occur with painful cramps. 1 , 21 Like patients with FFI, Morvan’s chorea also may manifest with hallucinatory behavior, a stuporous confused state, and peculiar motor disturbances, but these features are very rare in Morvan’s chorea. 1 , 22 Like the other subtypes of agrypnia excitata, polysomnograms in patients with Morvan’s chorea reveal the persistent absence of sleep rhythms for up to 4 months with the typical loss of slow-wave sleep. 1 , 21 Patients with Morvan’s chorea will exhibit REM without atonia and will also demonstrate hypercortisolemia and hypercatecholemia in the plasma. 2 , 21
 |
The exact pathophysiology of Morvan’s chorea remains uncertain. However, it is believed that the condition arises via autoimmune mechanisms. 21 The presence of voltage-gated potassium channel antibodies is also commonly found in the serum of patients with Morvan’s chorea. 20 , 21 , 23 The effect of these antibodies is believed to cause neuronal hyperexcitability by disrupting the ability of voltage gated potassium channel antibody to repolarize motor nerves effectively. 21 , 24
Unlike FFI, which runs a uniformly fatal course, Morvan’s chorea may resolve overtime but can, in some cases, result in death. 1 , 2 In severe cases encephalopathy, dementia, or reduplicative paramnesia may ensue. 2 , 20 , 23 Notwithstanding, given the potentially fatal course of other diseases that mimic Morvan’s chorea, consideration of a thorough differential diagnosis is critical to help minimize misdiagnosis. 25 Accurately recognizing Morvan’s chorea may help prevent misdiagnosis in the setting of clinically similar diseases such as in patients suffering from acquired forms of peripheral nerve hyperexcitability. Acquired peripheral nerve hyperexcitability can occur following timber rattlesnake envenomation or Guillain-Barre’ syndrome, two conditions that can lead to devastating sequelae. 22 , 25 Fortunately, Morvan’s chorea is, for the most part a treatable condition. 1 , 2 , 3 Unlike the other two components of agrypnia excitata, appropriate therapeutic approaches can reverse the course of Morvan’s chorea. 26 Plasmapheresis, thymectomy, and long-term immunosuppressive therapy can yield complete resolution of symptoms in patients with Morvan’s chorea. 20 , 26
Delirium Tremens
Delirium tremens describes the third and final element of agrypnia excitata. Delirium tremens often arises in the setting of sudden withdrawal in chronic alcoholics or individuals on barbiturates or benzodiazepines for extended periods. 19 , 27 Delirium tremens is a potentially fatal and debilitating disorder whose clinical features include delirium, visual and tactile hallucinations, formication, severe anxiety, and severe, involuntary tremors of the extremities. When occurring in the setting of agrypnia excitata, delirium tremens arises with an acute confused state associated with bizarre hallucinatory behavior, agrypnia, and motor and autonomic agitation. 1 , 27 The autonomic activation in agrypnia excitata-type delirium tremens may be so severe that it results in the death of the patient. 1 Polysomnogram evaluation of patients in delirium tremens reveals findings similar to those found in the other two components of agrypnia excitata. Polysomnogram recordings of delirium tremens patients demonstrate significantly reduced or completely absent slow-wave sleep in conjunction with sleep fragmentation and persistent nocturnal awakenings. 2 , 28 , 29 Furthermore, REM sleep without muscular atonia is also found in delirium tremens. 2 , 30
The degree of slow-wave sleep loss in all three forms of agrypnia excitata corresponds to the amount of atrophic change that occurs in the brain at the time of testing. 31 , 32 Similar to the other two components of agrypnia excitata, delirium tremens also demonstrates increased cortisol and catecholamines as well as tachypnea, irregular breathing, pyrexia, and hypertension throughout the day. 1 , 33 – 35 Although pathophysiologic mechanisms underlying delirium tremens remain unclear, deterioration of the thalamus and its related structures has been established as a primary factor in the development of the disorder. 1 , 36 – 38 Incidentally, imaging studies reveal significantly reduced activity in the thalamus of patients with delirium tremens. 39 The management of delirium tremens centers on supportive therapy in conjunction with pharmacologic management. The current mainstay of treatment includes benzodiazepines and mood-stabilizers until symptoms subside. 19 , 39
CONCLUSION
Agrypnia excitata represents a triad of three conditions that are characterized by insomnia, secondary drowsiness, and a stuporous mental state. The inability to sleep exacerbates the symptoms in such patients, leading to extreme exhaustion, coma, and ultimately death. Thalamic deterioration has been linked to the onset and course of agrypnia excitata and accounts for the typical clinical features of the disorder. The inability to generate sleep in such patients remains a primary challenge facing clinicians. Finally, a need exists for increased funding for controlled studies in order to offer improved insight into this devastating disease. In addition, these studies can improve current therapeutic approaches to agrypnia excitata and bring those individuals affected by the disease one step closer to living normal lives.
1. Montagna P, Lugaresi E: Agrypnia excitata: a generalized overactivity syndrome and a useful concept in the neurophysiopathology of sleep. Clin Neurophysiol 2002; 113:552–560Google Scholar
2. Lugaresi E, Provini F: Agrypnia excitata: clinical features and pathophysiological implications. Sleep Med Rev 2001; 5:313–322Google Scholar
3. Iriarte J, Ayuso T, Echavarri C, et al: Agrypnia excitata in fatal familial insomnia: a video-polygraphic study. Neurology 2007; 69:607–608Google Scholar
4. Lugaresi E, Tobler I, Gambetti P, et al: The pathophysiology of fatal familial insomnia. Brain Pathol 1998; 8:521–526Google Scholar
5. Montagna P, Gambetti P, Cortelli P, et al: Familial and sporadic fatal insomnia. Lancet Neurol 2003; 2:167–176Google Scholar
6. Spacey SD, Pastore M, McGillivray B, et al: Fatal familial insomnia: the first account in a family of chinese descent. Arch Neurol 2004; 61:122–125Google Scholar
7. Sundstrom DG, Dreher HM: A deadly prion disease: fatal familial insomnia. J Neurosci Nurs 2003; 35:300–305Google Scholar
8. Montagna P, Cortelli P, Avoni P, et al: Clinical features of fatal familial insomnia: phenotypic variability in relation to a polymorphism at codon 129 of the prion protein gene. Brain Pathol 1998; 8:515–520Google Scholar
9. Cortelli P, Perani D, Montagna P, et al: Pre-symptomatic diagnosis in fatal familial insomnia: serial neurophysiological and 18FDG-PET studies. Brain 2006; 129:668–675Google Scholar
10. Manetto V, Medori R, Cortelli P, et al: Fatal familial insomnia: clinical and pathologic study of five new cases. Neurology 1992; 42:312–319Google Scholar
11. Cortelli P, Gambetti P, Montagna P, et al: Fatal familial insomnia: clinical features and molecular genetics. J Sleep Res 1999; 8:23–29Google Scholar
12. AASM. The International Classification of Sleep Disorders. Revised and coding manual, 2nd ed. 2005:226Google Scholar
13. Schenkein J, Montagna P: Self management of fatal familial insomnia, part 1: what is FFI? MedGenMed 2006; 8:65Google Scholar
14. Schenkein J, Montagna P: Self-management of fatal familial insomnia, part 2: case report. MedGenMed 2006; 8:66Google Scholar
15. Dimitri D, Jehel L, Durr A, et al: Fatal familial insomnia presenting as psychosis in an 18-year-old man. Neurology 2006; 67:363–364Google Scholar
16. Waldman AD, Cordery RJ, MacManus DG, et al: Regional brain metabolite abnormalities in inherited prion disease and asymptomatic gene carriers demonstrated in vivo by quantitative proton magnetic resonance spectroscopy. Neuroradiology 2006; 48:428–433Google Scholar
17. Piao YS, Kakita A, Watanabe H, et al: Sporadic fatal insomnia with spongiform degeneration in the thalamus and widespread PrPSc deposits in the brain. Neuropathology 2005; 25:144–149Google Scholar
18. Pandya M, Pozuelo L, Malone D: Electroconvulsive therapy: what the internist needs to know. Cleve Clin J Med 2007; 74:679–685Google Scholar
19. DeBellis R, Smith BS, Choi S, et al: Management of delirium tremens. J Intensive Care Med 2005; 20:164–173Google Scholar
20. Hudson LA, Rollins YD, Anderson CA, et al. Reduplicative paramnesia in Morvan’s syndrome. J Neurol Sci 2008; 267:154–157Google Scholar
21. Liguori R, Vincent A, Clover L, et al: Morvan’s syndrome: peripheral and central nervous system and cardiac involvement with antibodies to voltage-gated potassium channels. Brain 2001; 124:2417–2426Google Scholar
22. Newsom-Davis J, Buckley C, Clover L, et al: Autoimmune disorders of neuronal potassium channels. Ann N Y Acad Sci 2003; 998:202–210Google Scholar
23. McKeon A, Marnane M, O’Connell M, et al. Potassium channel antibody associated encephalopathy presenting with a frontotemporal dementia like syndrome. Arch Neurol 2007; 64:1528–1530Google Scholar
24. Sinha S, Newsom-Davis J, Mills K, et al: Autoimmune aetiology for acquired neuromyotonia (Isaacs’ syndrome). Lancet 1991; 338:75–77Google Scholar
25. Gutmann L, Gutmann L: Axonal channelopathies: an evolving concept in the pathogenesis of peripheral nerve disorders. Neurology 1996; 47:18–21Google Scholar
26. Haug BA, Schoenle PW, Karch BJ, et al: Morvan’s fibrillary chorea: a case with possible manganese poisoning. Clin Neurol Neurosurg 1989; 91:53–59Google Scholar
27. Tachibana M, Tanaka K, Hishikawa Y, et al: A sleep study of acute psychotic states due to alcohol and meprobamate addiction, in Advances in Sleep Research. Edited by Weitzman ED. New York, Spectrum, 1975, pp 177–205Google Scholar
28. Brower KJ: Alcohol’s effects on sleep in alcoholics. Alcohol Res Health 2001; 25:110–125Google Scholar
29. Gann H, Feige B, Hohagen F, et al: Sleep and the cholinergic rapid eye movement sleep induction test in patients with primary alcohol dependence. Biol Psychiatry 2001; 50:383–390Google Scholar
30. Schenck CH, Bundlie SR, Ettinger MG, et al: Chronic behavioral disorders of human REM sleep: a new category of parasomnia. Sleep 2002; 25:293–308Google Scholar
31. Ishibashi M, Nakazawa Y, Yokoyama T, et al: Cerebral atrophy and slow wave sleep of abstinent chronic alcoholics. Drug Alcohol Depend 1987; 19:325–332Google Scholar
32. Imatoh N, Nakazawa Y, Ohshima H, et al: Circadian rhythm of REM sleep of chronic alcoholics during alcohol withdrawal. Drug Alcohol Depend 1986; 18:77–85Google Scholar
33. Iranmanesh A, Veldhuis JD, Johnson ML, et al: 24-hour pulsatile and circadian patterns of cortisol secretion in alcoholic men. J Androl 1989; 10:54–63Google Scholar
34. Risher-Flowers D, Adinoff B, Ravitz B, et al: Circadian rhythms of cortisol during alcohol withdrawal. Adv Alcohol Subst Abuse 1988; 7:37–41Google Scholar
35. Clark LT, Friedman HS: Hypertension associated with alcohol withdrawal: assessment of mechanisms and complications. Alcohol Clin Exp Res 1985; 9:125–130Google Scholar
36. Kopelman MD: The Korsakoff syndrome. Br J Psychiatry 1995; 166:154–173Google Scholar
37. Pagnan L, Berlot G, Pozzi-Mucelli RS Magnetic resonance imaging in a case of Wernicke’s encephalopathy. Eur Radiol 1998; 8:977–980Google Scholar
38. Shimamura AP, Jernigan TL, Squire LR: Korsakoff’s syndrome: radiological (CT) findings and neuropsychological correlates. J Neurosci 1988; 8:4400–4410Google Scholar
39. Wang GJ, Volkow ND, Franceschi D, et al: Regional brain metabolism during alcohol intoxication. Alcohol Clin Exp Res 2000; 24:822–829Google Scholar

