
The Neuropsychiatry of Chronic Cocaine Abuse
Abstract
This review integrates findings from neuropsychological, PET, and MRI studies in human subjects and neurochemical findings in animals to make inferences about neuropsychiatric consequences of chronic abuse of cocaine. It also aims to develop insights into brain–behavioral relationships that may explain the perpetuation of addictive behaviors. Such insights promise to lead to a better understanding of the neuropsychiatry of cocaine abuse and to promote the development of more efficacious treatments. The authors present evidence suggesting that cocaine abusers have specific dysfunction of executive functions (decision making, judgment) and that this behavior is associated with dysfunction of specific prefrontal brain regions, the orbitofrontal cortex, and anterior cingulate gyrus. Suggestions for future research and treatment are also discussed.
The first known use of cocaine occurred between 2000 and 1500 b.c. among South American Indians. From the 1920s to the 1960s, the drug was used recreationally in the United States among jazz musicians, actors, and the “cultural avant garde.”1 Since the early 1970s, cocaine use has grown dramatically in popularity. Estimates suggest that the number of Americans who have tried cocaine at least once rose from 5.4 million in 1974 to 21.6 million in 1982. Indeed, cocaine production has come to flourish as an industry, with an estimated annual revenue of at least 27 billion dollars. In 1985, the introduction of “crack” brought a subsequent increase of cocaine use; a study published in 1988 estimated 5,000 new users per day, 6 million regular users, and 1 million compulsive users of the drug.2 In 1995, an estimated 1.5 million Americans used cocaine, and around a half-million used cocaine at least weekly.
Obviously, this staggering number of cocaine users catapulted cocaine abuse to a major public health problem. Compulsive, life-threatening behaviors are often observed in humans who abuse this drug. The chronic use of cocaine is also associated with major medical, neurological, and neuropsychiatric complications. Moreover, there is at present no adequate therapy for cocaine addiction. It is likely that the inadequacy of therapeutic approaches reflects the lack of a neurobiologic model of cocaine abuse that takes into consideration the neurological sequelae of repeated cocaine use.3
In this review we address this issue by focusing attention on recent findings in studies of brain function and integrity as related to cocaine abuse in humans. We also describe a working hypothesis that prefrontal lobe damage is the pathological substrate that must be better understood before efficient therapy can be developed for individuals addicted to cocaine. We suggest that complex interactions of cortical and subcortical brain regions are involved in the maintenance of cocaine addiction—and that therefore pharmacological approaches directed to one neurotransmitter might not necessarily be the most productive way to deal with cocaine abuse.
CHRONIC EFFECTS OF COCAINE ON MONOAMINERGIC SYSTEMS
Although activation of midbrain dopamine (DA) neurons is thought to play the most important role in appetitive behavior,4 studies in animals suggest that chronic administration of cocaine causes alterations in dopamine dynamics in various brain regions.5–7 Studies of cerebral glucose metabolism suggest that the main effects of cocaine primarily involve subcortical dopaminergic circuits in rats,8,9 but cortical involvement, possibly including serotonergic and noradrenergic systems, is evidenced in the nonhuman primate after chronic treatment.10 Although DA concentrations have not been measured directly in the brains of abstinent cocaine abusers, a relationship was found between decreased D2 dopamine receptor availability and decreased glucose metabolism in several frontal brain regions, particularly the orbitofrontal cortex and cingulate gyrus,11 which receive dopaminergic afferents. Decreased D2 receptor availability also was correlated with years of cocaine use. Volkow et al.11 suggested that decreased D2 receptor availability could reflect downregulation of postsynaptic D2 receptors in response to repeated elevations in synaptic DA when cocaine is administered. An association between depletion of DA and decreased frontal metabolism also has been observed in Parkinson's disease.12 Postmortem assays of brains from human subjects with no known neurological or psychiatric disease reveal that cocaine binds to dopamine uptake sites in the basal ganglia, but also to serotonin uptake sites in the frontal cortex and hippocampus.13 It is also important to point out that the number of DA transporters is decreased in the prefrontal cortex of cocaine users.7
Monoamine (dopamine and serotonin) depletion or dysregulation in specific brain regions that control drive and affect could contribute to neuropsychiatric abnormalities (craving and depression) evident in the aftermath of chronic cocaine abuse.11,14,15 The idea that chronic cocaine administration in humans might result in dopamine depletion is supported by reports of hyperprolactinemia14 as well as increased pituitary volume16 in cocaine abusers. Furthermore, individuals withdrawing from cocaine retrospectively report craving that is correlated with elevated glucose metabolism in the orbitofrontal cortex (OFC) and other prefrontal areas.17 Therefore, the repeated use of cocaine may result in cocaine-induced dopaminergic stimulation, and shortly after cessation of drug administration, activation in the OFC and other prefrontal brain regions may lead to increased drive to self-administer cocaine compulsively. With continued abstinence from cocaine use, these regions become hypometabolic, but when cocaine is readministered, frontal brain regions may be reactivated, again contributing to the compulsion to use cocaine.18
Chronic cocaine users have reported what we have called cocaine withdrawal–related depression,3 which may be a neurophysiologic response to the elimination of cocaine from the CNS. This could occur according to the following scenarios. For example, the use of cocaine is associated with acute increases in dopamine levels in the synaptic cleft and overstimulation of postsynaptic DA receptors; the chronic stimulation of these receptors could lead to a new adaptive state such as receptor downregulation or dysregulation. Therefore, continuous use of the drug would be required to sustain the new biochemical steady state in the CNS. On the other hand, repeated use of the drug may alter the dynamics of monoamine turnover as a response to the repeated cocaine blockade of DA reuptake. This could occur via GABAergic striatonigral feedback pathways that could act to dampen the synthesis and release of monoamines after chronic exposure to cocaine. It is also likely that chronic use of cocaine, by causing upregulation of presynaptic DA transporters,19 would decrease the amount of DA available to stimulate postsynaptic receptors because of increased DA reuptake by increased numbers of membrane DA transporters. If any of these scenarios is correct, then the addict would need to use cocaine repeatedly in order to maintain homeostasis within central dopaminergic systems.
EFFECTS OF COCAINE ON REGIONAL AND GLOBAL BRAIN FUNCTION
Cerebral Blood Flow and Glucose Metabolism as Indices of Brain Function
Measures of global and regional cerebral blood flow (CBF and rCBF, respectively) are indices of cerebral functional activity. CBF is regulated according to the requirements of cerebral metabolism.20 In animals, in normal human subjects, and in patients with brain disease,21 rates of rCBF are related to regional cerebral metabolic rates for glucose,22 and in some cases (during hard exercise), rCBF is coupled to the regional cerebral metabolic rate for oxygen in the cerebral cortex.23 Cerebral blood flow falls in neuropathologic states, including dementia24 and stroke.25 Therefore, measurements of CBF and rCBF provide information about functional brain activity. Positron emission tomography (PET), a noninvasive nuclear medicine technique, makes it possible to address questions about functional brain activity in humans. This technique can be used to quantify CBF, rCBF, and cerebral metabolic rates for glucose, during either rest or cognitive activation.
Animal Studies
Acute administration of cocaine in rodents and primates has profound metabolic effects on the ventral striatum.10,26 It increases cerebral metabolism in rats, primarily in subcortical regions,8,26 whereas in nonhuman primates, glucose utilization is decreased in the limbic system, including the nucleus accumbens, limbic cortex, orbitofrontal and other prefrontal cortical areas, and part of the hippocampus formation.10 This interspecies difference in the effects of cocaine on brain metabolism may be attributed to more complex neuroanatomical architecture in the primate as compared with the rodent. The results suggest that in the primate, the central action of cocaine involves corticostriatal activity, originating in limbic cortex and projecting to the ventral striatum. It is through these projections that the reinforcing effects of cocaine may be mediated, in part, by limbic cortical structures.10 The discrepancy in findings between rodent and primate studies illustrates the limitations of animal models of chronic cocaine use and justifies the need to investigate the chronic effects of cocaine in humans.
Human Studies
Few studies have assessed the acute effects of cocaine because of the difficulties inherent in administering cocaine to humans. However, cerebral glucose utilization, as measured by PET and [18F] fluorodeoxyglucose, was reduced when cocaine was administered to volunteers with histories of intravenous cocaine abuse.27 Significant decreases were found in most brain regions assayed. Thus, cocaine-induced decreases in metabolism are similar in humans and nonhuman primates. Normalized cerebral blood flow, as assessed by single-photon emission computed tomography (SPECT), was also decreased in the frontal cortex and basal ganglia and was negatively correlated with increases in self-ratings of “high” and “rush.”28
In abstinent heavy cocaine abusers, cerebral metabolism is higher in medial orbitofrontal cortex and basal ganglia during the first week of abstinence from cocaine as compared with metabolism in normal subjects, but then falls below normal levels and remains depressed for at least 3 months.11,17,29 In cocaine abusers abstinent 1 to 6 weeks, glucose utilization also correlated mildly with an estimate of weekly dose of cocaine and years of cocaine use.29 Although interesting, these latter results should be interpreted cautiously because they are based on only 7 patients. Nevertheless, the relationship between intensity of cocaine use and low level of frontal lobe metabolism suggests that the heavy use of cocaine is at least partly responsible for these findings. In contrast, Stapleton et al.30 did not show frontal hypometabolism in chronic cocaine abusers who also used opiates (polysubstance abusers). This discrepancy is probably due to differences in the amount of cocaine used. The cocaine abusers studied by Volkow et al.29 used larger amounts of cocaine than those participants studied by Stapleton et al.30 (>4 g/week versus about 1.4 g/week) and they lacked histories of illicit opiate abuse. In addition, the users in Volkow et al.29 used crack, and the route of administration was via smoking, whereas the users in Stapleton et al.30 used multiple methods of administration.
Studies of rCBF also show defects in prefrontal cortex in abstinent cocaine abusers.15,31,32 These rCBF deficits could reflect long-term vasospasm in cerebral arteries exposed chronically to the sympathomimetic actions of cocaine.15 Greater deficits are observed in rCBF than in glucose utilization when both are examined in the same cocaine-abusing individual.15 The discrepancy may reflect the direct actions of cocaine on cerebral vessels. Frontal lobe deficits in rCBF may be related to selective sensitivity of anterior and middle cerebral arteries to cocaine.
Changes in cerebral perfusion may be related to changes in neurocognitive functioning. Strickland et al.33 reported an association between hypoperfusion and neurocognitive difficulties in the areas of attention/concentration, new learning, visual and verbal memory, word production, and visuomotor integration. However, only 8 subjects were studied 6 months after their last reported use of cocaine. Therefore, it is uncertain whether the changes in rCBF and neurobehavioral functioning can be attributed to cocaine use. Other studies find minimal neurocognitive symptoms in cocaine abusers who show decreased cerebral blood flow.15,32 However, these studies failed to present detailed results of the specific tests used. Discrepant results may reflect differences in demographic characteristics (personality characteristics, amount/duration of cocaine use), sensitivity of the neurobehavioral tests, and research methodology.
CEREBROVASCULAR EFFECTS OF COCAINE
With the advent of freebase cocaine (crack) in 1985, the number of reports of cocaine-related ischemic and hemorrhagic strokes increased dramatically.34–38 Underlying aneurysms or arteriovenous malformations (AVMs) are associated with hemorrhagic strokes. Cocaine-induced vasoconstriction, on the other hand, appears to cause ischemic strokes. The vasoconstrictive action of cocaine and the cocaine metabolites benzoylecgonine39 and norcocaine40 may be the greatest contributing factor to the increased incidence of ischemic strokes in cocaine users. Amphetamine, which is pharmacologically similar to cocaine in terms of its effects on dopamine levels, produces vasoconstriction on small arterial branches of the anterior and middle cerebral arteries when intravenously administered to monkeys.41 If cocaine were to affect the same vessels, then brain regions such as the frontal and prefrontal cortices, which are in the distribution of these vessels, would be most affected by repeated use of cocaine. Vasoconstriction might follow from the increased availability of epinephrine and norepinephrine in the vasculature due to cocaine blockade of reuptake. Serotonin might also play a role because it is a potent vasoconstrictor of large and medium-sized brain vessels.35,42 Other possible mechanisms for cerebral ischemia associated with cocaine abuse include hypertension, platelet aggregability, vasculitis, cardiogenic emboli, and drug contaminants.43,44 Intravenous cocaine administration increases the risk of hemorrhagic stroke.36,45 Although stroke may be more likely to occur in cocaine abusers who have preexisting risk factors for stroke (AVMs, vascular disease, cardiac sources of emboli), the presence of an underlying structural lesion is not a necessary factor for stroke. In these cases, sudden increases in arterial pressure could be enough to cause hemorrhagic strokes.
EFFECTS OF COCAINE ON BRAIN STRUCTURE
Available information on the effects of chronic cocaine use on brain structure is limited. However, such abnormalities have been found in chronic cocaine abusers46,47 and polysubstance abusers.48 When Pascual-Leone et al.46 compared planimetric CT measurements of habitual cocaine abusers with those of first-time users and control subjects, habitual users had a greater degree of cerebral atrophy, which correlated with duration of cocaine abuse on one measure (maximal frontal horn width).46 However, no measurement was made of amount or intensity of cocaine use. In addition, the study included participants with histories of seizures, acute psychosis, and tics. Therefore, it is possible that the cerebral atrophy was related to long-standing psychopathology rather than cocaine use. Langendorf et al.47 used volumetric MRI measures to study brain atrophy in individuals who used cocaine alone. Their preliminary results suggest reduced brain volume in cocaine abusers. These authors suggest that cerebral ischemia is the pathogenesis for atrophy and that this ischemia may be partially reversible with abstinence from cocaine.
Studies from our laboratories have found variable results, the variability probably depending on the technique used for measurement and the level of use of cocaine and other drugs in the group under investigation. Cascella et al.48 used planimetric CT and found no relationship between a measure of brain atrophy and cocaine use once the effect of age was controlled. This study group included polysubstance abusers who used on average less than 2 grams per week of cocaine. In another study, ventricle-to-brain ratio (VBR) was determined volumetrically by MRI in polysubstance abusers who had a median value of less that one-half gram of cocaine use per week, and no group differences were found in VBR.49 However, in our latest endeavor, we found significantly smaller total volume of the prefrontal lobe (left and right hemispheres) in a polysubstance abuse group relative to control subjects (Liu et al., submitted for publication). The drug abusers had a median value of about a gram of cocaine use per week. These data suggest that it is possible to detect differences in brain structure in chronic drug users if a sensitive measure of brain morphology is used and if the amount of cocaine use is high enough to cause changes in structure.
EFFECTS OF COCAINE ON NEUROBEHAVIORAL FUNCTIONING
The term neurobehavioral effects includes alterations in cognition, personality, affect, and behavior. Findings on the neurocognitive effects of chronic cocaine abuse are equivocal. Some studies report difficulties with response speed, visual and verbal learning and memory, and executive functions (Bolla et al., unpublished data),33,50–55 while others report minimal effects.29,32,56 Interestingly, Manschreck et al.52 found an association between chronic cocaine use and enhancement of performance on some tests (e.g., finger tapping, visuospatial memory, and learning). These discrepancies most likely arise from differences in the amount and/or duration of cocaine use, the length of abstinence when tested, the sensitivity of the neuropsychological tests given, the lack of a control group, small sample size, and/or poor control of severity of depression, age, premorbid level of intelligence, gender distribution, and personality characteristics (e.g., antisocial personality disorder).
When interpreted collectively, studies on the neurocognitive effects of chronic cocaine abuse suggest that long-term neurologic sequelae of chronic cocaine abuse may be subtle and specific rather than general. Problems with executive functioning (decision making, judgment, attention/planning/mental flexibility) are the most frequently reported cognitive difficulties. This cognitive domain relates primarily to the functional integrity of the prefrontal lobe.57–59
Chronic cocaine abuse may also lead to persistent changes in brain function and structure with no detectable neurocognitive effects. Few neurocognitive effects were found in cocaine abusers with decreased cerebral blood flow.1 ,32,56 Compensatory increases in the rate of oxygen extraction under conditions of decreased CBF may explain this finding.60 Nonetheless, neurobehavioral tests have rarely been used to study chronic neurobehavioral effects of cocaine. Detecting subtle behavioral effects requires the administration of several complex tests of executive functioning. Also, tests measuring the accuracy and speed of information processing have proved to be the most sensitive in detecting subtle brain dysfunction. Such tests have not been used routinely.
Although the possibility exists that cocaine abuse might stem from preexisting cognitive deficits, evidence that cocaine use contributes, at least partly, to the development of cognitive difficulties comes from studies showing negative relationships between test scores, amount and duration of cocaine abuse, and length of abstinence (Bolla et al., unpublished data).53,55
Changes in affect and personality have also been associated with chronic cocaine abuse. For example, the depressed mood reportedly associated with cocaine withdrawal may also account for a portion of the cognitive difficulties found in chronic cocaine abusers.54,61 Antisocial personality disorder has frequently been diagnosed in cocaine abusers and has also been associated with frontal lobe dysfunction. This relationship has been demonstrated by neuropsychological testing62 and the use of event-related potentials.63 Thus, affect and personality characteristics must also be considered when attempting to characterize the neurologic effects of chronic cocaine abuse.
GENERAL DISCUSSION
The accumulated evidence apparently supports the view that chronic cocaine abuse is associated with abnormalities in the central nervous system. More specifically, neurocognitive deficits are often reported in executive functions (judgment, decision making, mental flexibility) of chronic cocaine abusers. In addition, imaging studies (SPECT and PET) have revealed perfusion deficits and metabolic changes in the limbic prefrontal cortex of cocaine abusers. The limbic prefrontal brain region is an important anatomical substrate of executive cognitive abilities. Orbitofrontal and anterior cingulate cortices may be particularly important to study because previous neurochemical, neurobehavioral, and neuroimaging investigations have reported alterations in these regions in chronic cocaine abusers.
Chronic abusers of cocaine exhibit lack of judgment, unreliability, poor foresight, difficulty making decisions, disinhibition, apathy, euphoria, and irritability. The prefrontal lobe appears to play a role in the control or modulation of these behaviors, since damage to this region produces similar behaviors in brain-injured patients who are not drug users.57,64 Therefore, it is logical to suggest that the behavioral syndrome associated with the long-term use of cocaine may be associated with functional changes in the prefrontal cortex.
Because prefrontal brain regions are also involved in the regulation of drive and control, their dysfunction could lead to an inability to abstain from the use of cocaine. In primates, the orbitofrontal cortex is involved in positive reinforcement,65 and neurons in this region increase their firing rate during rewarding electrical stimulation to other brain regions.66,67 The OFC is also involved, at least to some extent, in both spontaneous and cue-elicited craving. A correlation was observed between retrospective reports of spontaneous cocaine craving and changes in metabolic activity in the orbitofrontal cortex.17 Although the presentation of cocaine-related cues increased the metabolic rate for glucose in the OFC, these changes in metabolism were not correlated with cue-elicited craving.68 Cocaine abuse is also associated with repetitive compulsive acts that perpetuate drug-seeking behaviors. It is notable that patients who suffer from obsessive-compulsive disorder have metabolic abnormalities in the OFC.69,70 Therefore, the available evidence suggests that the OFC may be an important player in the maintenance of addiction.
The cingulate cortex may be another significant anatomical region in the developmental process of addiction. The cingulate cortex, like the OFC, has been shown to be involved in positive reinforcement in animal studies.71 The anterior cingulate cortex may also participate in the modulation of the execution of appropriate responses and the suppression of inappropriate response.72 Additionally, a “memory” of the positive drug experience may contribute to perpetuation of use and may lead to a feeling of craving when the drug is discontinued after the repeated administration of cocaine.73 The exact neurobiologic substrates for consolidation of this “memory” are unknown but probably involve the cingulate gyrus and other brain regions.
To determine how abnormalities in brain structure affect brain function and neurocognitive functioning, it is necessary to examine relationships among volumetric changes and rCBF and neurobehavioral performance. This approach was successfully used to extend knowledge about the effects of alcohol on the CNS, and a similar approach might be useful to study the neurologic effects of cocaine. Using multiple measures of CNS functioning (glucose metabolism, MRI, and neurobehavioral testing), Wang et al.74 showed that cortical atrophy (measured by quantitative MRI), but not ventricular size, was related to global decreases in brain glucose metabolism of alcoholics. However, in both alcoholics and control subjects, cortical atrophy and ventricular enlargement were both associated with decreased metabolism predominantly in the frontal cortices and subcortical structures. In addition, MRI findings (degree of ventricular enlargement and brain atrophy) were correlated with frontal lobe metabolism but not with neuropsychological performance. This relationship suggests that mild structural changes coupled with metabolic deficits alone may not be enough to produce clinically relevant neurobehavioral deficits.
A MODEL OF THE CLINICAL NEUROBIOLOGICAL EFFECTS OF CHRONIC COCAINE ABUSE
The following neurobehavioral model of cocaine effects (illustrated in Figure 1) is presented to summarize the neurologic effects of acute and chronic cocaine abuse. At a basic level, acute and chronic cocaine use alters neurotransmission in the central nervous system. Monoamine (dopamine and serotonin) depletion or neurochemically mediated dysregulation of specific brain regions (prefrontal cortex, limbic system) has been associated with alterations in cerebral blood flow and glucose metabolism. The prefrontal cortex and limbic system are involved in drive and affect. Thus, alterations in these brain structures could contribute to the reinforcing and addictive properties of cocaine. Alterations in neurotransmission, blood flow, and metabolism can also result in neurobehavioral effects (cognitive deficits; psychiatric effects including depression and compulsive behavior). Cocaine-induced neurofunctional changes may contribute to the repetitive and compulsive behavioral patterns associated with addictive behaviors. Although a one-to-one correspondence among neurotransmitter, functional, and behavioral changes probably does not exist, drug abusers who develop clinically relevant neurobehavioral changes will likely show cerebral blood flow changes in PET studies. The repeated vasoconstrictive actions of cocaine and the purported ischemic changes that this vasoconstriction might cause could result in progressive cell death due to reoxygenation-induced injury through oxidative and excitotoxic mechanisms.
This discussion predicts that various neurologic impairments may result from chronic cocaine abuse. A number of neurologic abnormalities have indeed been reported in association with cocaine abuse. These include cerebrovascular events (strokes), vasculitis, and seizures, as well as alterations in brain structure, metabolism, and blood flow. Nevertheless, the possibility must be considered that some of these neurologic impairments may predate cocaine use. It should be pointed out, however, that the determination of cause may be irrelevant to the development of treatment strategies that will target the clinical manifestations of a neuropathologic state. For example, although levodopa replacement has been very successful in the treatment of Parkinson's disease, the cause of dopaminergic cell death still remains to be determined.
FUTURE DIRECTIONS FOR RESEARCH AND TREATMENT
Future research will be needed to determine if chronic cocaine abuse is associated with changes in neurocognitive performance, cerebral blood flow, and structure. Such findings would elucidate brain–behavior relationships in cocaine-abusing individuals. An understanding of these relationships in individuals with cocaine addiction will lead to more effective treatment of cocaine dependence.
To some extent, controversy about the neurological sequelae of chronic cocaine abuse reflects a lack of rigor in measurement and examination of level of exposure. In the field of clinical neurotoxicology, when occupationally exposed workers were compared with a group of control subjects, few difficulties were found in neurocognitive performance. However, when the exposed group was subdivided with respect to exposure (high, medium, low), group differences became evident. These differences may exist because the effects of chemicals on the CNS are generally subtle and may occur only at higher doses. Therefore, it is critical to compare individuals with the highest exposure to individuals with lower or no exposure. This logic needs to be generalized to the study of the effects of chronic cocaine on the CNS. Clinical neurotoxicology principles predict that only those with high cocaine use might show neurologic deficits after cocaine use. Indeed, this approach has been successful in showing that the degree of cocaine use was proportional to the magnitude of performance difficulties (Bolla et al., unpublished data).53,55
It is also imperative to collect the correct “exposure” measure for analysis. Measures that have been used are intensity (amount), duration, and total dose (amount×duration). Selection of the correct parameter depends on the toxicokinetics of the chemical/drug. For example, exposure duration was critical to the development of neurocognitive effects after organic and inorganic lead exposure,75 whereas exposure intensity was more critical to the development of neurocognitive effects following exposure to organic solvents.76 Cascella et al.48 also found that amount of alcohol consumption per session at the time of peak use (bingeing) was a better predictor of brain atrophy than current alcohol consumption. Therefore, it is imperative to collect information on intensity (grams/week), duration (months), peak use, and total dose (intensity×duration) of cocaine and other drugs used in order to determine which “exposure” measure is critical to changes in brain structure and function. Our preliminary work found a stronger negative relationship between neurobehavioral performance and intensity of cocaine use than between neurobehavioral performance and duration of use. These results are similar to observations related to the neurotoxic effects of amphetamine analogs, where repeated administration of small doses in one day can cause more lasting effects on brain monoaminergic systems than single injections of the drugs over long periods of time.77
Although previous work shows abnormalities in cerebral blood flow and metabolism in abstinent cocaine abusers, few have attempted to directly examine the behavioral correlates of these alterations. In addition, to our knowledge no investigator has used a cognitive activation task during PET acquisition to increase the sensitivity of detection of subtle brain changes. Studies of this type are critical to identifying the neurobiological bases of behavioral deficits seen in cocaine abusers.
Critically examining the relationship between neurobehavioral and rCBF correlates requires measurement of rCBF while the cocaine abuser is engaged in a cognitive activity. In activation studies, changes in rCBF are measured while an individual is performing a cognitive task. Activation studies may increase the sensitivity as well as the specificity of PET.78 Cognitive tasks designed to selectively activate the OFC and ACC would yield important information on the functional integrity of these brain regions in cocaine abusers. Activation studies have been used to study brain function in normal volunteers,79–81 patients with Alzheimer's disease, and patients with schizophrenia,82 but there are no published activation studies on chronic cocaine abusers.
Cerebrovascular effects of cocaine could lead to structural abnormalities and associated clinically significant changes in behavior. To extend our knowledge about the chronic effects of cocaine on the CNS, studies with MRI are needed to measure regional brain volumes. Functional correlates (rCBF and neurocognitive measures) of these possible structural abnormalities could also be investigated in the same individuals. Approaches using multiple measures of brain integrity have been successful in the study of alcoholics74 and need to be extended to users of other drugs. Our model predicts a correlation between changes in brain volume and changes in rCBF. It is possible that these changes will be most prominent in the OFC and anterior cingulate gyrus and that they will be proportional to the intensity and/or duration of cocaine use. Furthermore, the model predicts that the magnitude of structural changes might determine the degree of neurobehavioral performance deficits. It is highly likely that these deficits will be in executive functions, since dysfunction in executive abilities may result in the perpetuation of addictive behavior. Deficits in executive functions can make it difficult to self-monitor, to change inappropriate behaviors, and to make rational decisions. Hence, a chronic cocaine abuser with these deficits would likely fail in attempts to discontinue self-destructive, drug-seeking behavior.
Our model also predicts that chronic cocaine abuse will be associated with lower performance on neurocognitive tests that measure prefrontal lobe functioning (executive functioning) and that the magnitude of performance difficulty will be correlated negatively with intensity of cocaine use. Also, the severity of these deficits should be proportional to the severity of the rCBF and structural (MRI) changes.
The use of a multi-method approach that examines the functional (rCBF) and structural (brain volume) correlates of neurobehavioral effects will provide valuable information about the neurobiology of chronic cocaine abuse. A better understanding of brain–behavior relationships will ultimately lead to more effective treatment and vocational rehabilitation of the chronic cocaine abuser. For example, specific pharmacological agents that normalize functional alterations (such as prefrontal hypometabolism) could be used in conjunction with specific cognitive-behavioral treatment programs. These customized treatment plans would take into account, and help the drug abuser compensate for, specific cognitive processing difficulties. In addition, knowledge of strengths in other cognitive domains could ultimately guide vocational rehabilitation programs to integrate these individuals into the workforce.
SUMMARY
In this review we have attempted to integrate the current state of knowledge on the biochemistry, cerebral blood flow, brain structure, and neurocognitive behavior of cocaine abusers. This discussion has reemphasized the view that chronic addiction is associated with brain disorders.3 Ideally, this information can help in developing hypothesis-driven and neurobiologically based treatments for cocaine addiction. The presentation of chronic cocaine abuse as a neuropsychiatric syndrome opens up avenues for future research focused on improvement of patient care. Emphasis on the clinical neurosciences of drug abuse can also lead to a redefinition of addiction using more appropriate biomedical/neurobiological models. These new approaches should take into consideration persistent changes in the functional neuroanatomy of brains chronically exposed to cocaine.
ACKNOWLEDGMENTS
This work was supported by the Intramural Research Program of the National Institute on Drug Abuse.
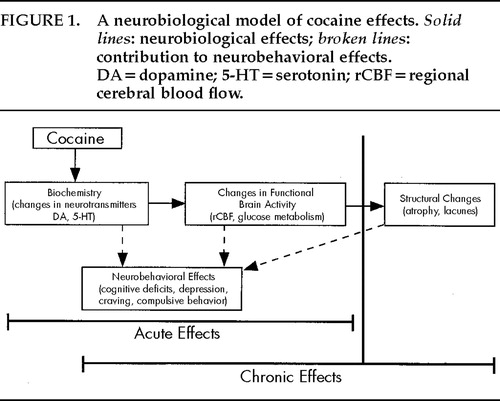
FIGURE 1. A neurobiological model of cocaine effects. Solid lines: neurobiological effects; broken lines: contribution to neurobehavioral effects. DA=dopamine; 5-HT=serotonin; rCBF=regional cerebral blood flow.
1. Peterson RC: Cocaine: an overview, in NIDA Research Monograph 13, edited by Stillman RC, Peterson RC. Washington, DC, DHEW, 1977, pp 17–34Google Scholar
2. Barnes DM: Drugs: running the numbers. Science 1988; 240:1729–1731Crossref, Medline, Google Scholar
3. Cadet JL, Bolla KI: Chronic cocaine use as a neuropsychiatric syndrome: a model for debate. Synapse 1996; 22:28–34Crossref, Medline, Google Scholar
4. Mirenowicz J, Schultz W: Preferential activation of midbrain dopamine neurons by appetitive rather than aversive stimuli. Nature 1996; 379:449–451Crossref, Medline, Google Scholar
5. Kuhar MJ, Pilotte NS: Neurochemical changes in cocaine withdrawal. Trends Pharmacol Sci 1996; 7:260–264Crossref, Google Scholar
6. Wilson JM, Levey AI, Bergeron C, et al: Striatal dopamine, dopamine transporter and vesicular monoamine transporter in chronic cocaine users. Ann Neurol 1996; 40:428–439Crossref, Medline, Google Scholar
7. Hitri A, Casanova MF, Kleinman JE, et al: Fewer dopamine transporter receptors in the prefrontal cortex of cocaine users. Am J Psychiatry 1994; 151:1074–1076Crossref, Medline, Google Scholar
8. London ED, Wilkerson G, Goldberg SR, et al: Effects of l-cocaine on local cerebral glucose utilization in the rat. Neurosci Lett 1986; 68:73–78Crossref, Medline, Google Scholar
9. Porrino LJ, Domer FR, Crane AM, et al: Selective alterations in cerebral metabolism within the mesocorticolimbic dopaminergic system produced by acute cocaine administration in rats. Neuropsychopharmacologia 1988; 1:109–118Crossref, Medline, Google Scholar
10. Lyons D, Friedman DP, Nader MA, et al: Cocaine alters cerebral metabolism within the ventral striatum and limbic cortex of monkeys. J Neurosci 1996; 16:1230–1238Crossref, Medline, Google Scholar
11. Volkow ND, Fowler JS, Wang GJ, et al: Decreased dopamine D2 receptor availability is associated with reduced frontal metabolism in cocaine abusers. Synapse 1993; 14:169–177Crossref, Medline, Google Scholar
12. Mayberg HS, Starkstein SE, Sadzot B, et al: Selective hypometabolism in the inferior frontal lobe in depressed patients with Parkinson's disease. Ann Neurol 1990; 28:57–64Crossref, Medline, Google Scholar
13. Biegon A, Dillon KA, Volkow ND, et al: Quantitative autoradiography of cocaine binding sites in human brain postmortem. Synapse 1992; 10:126–130Crossref, Medline, Google Scholar
14. Dackis CA, Gold MS: New concepts in cocaine addiction: the dopamine depletion hypothesis. Neuroscience and Biobehavior Review 1985; 9:469Crossref, Medline, Google Scholar
15. Volkow ND, Mullani H, Gould KL, et al: Cerebral blood flow in chronic cocaine users: a study with positron emission tomography. Br J Psychiatry 1988; 152:641–648Crossref, Medline, Google Scholar
16. Teoh SK, Mendelson JG, Woods BT, et al: Pituitary volume in men with concurrent heroin and cocaine dependence. J Clin Endocrinol Metab 1993; 76:1529–1532Medline, Google Scholar
17. Volkow ND, Fowler JS, Wolf AP, et al: Changes in brain glucose metabolism in cocaine dependence and withdrawal. Am J Psychiatry 1991; 148:621–626Crossref, Medline, Google Scholar
18. Volkow ND, Fowler JS: Brain-imaging studies of the combined use of cocaine and alcohol and of the pharmacokinetics of cocaethylene. NIDA Research Monograph 1994; 138:41–56Medline, Google Scholar
19. Tella SR, Landenheim B, Andrews AM, et al: Differential reinforcing effects of cocaine and GBR-12909: biochemical evidence for divergent neuroadaptive changes in the mesolimbic dopaminergic system. J Neurosci 1996; 16:7416–7427Crossref, Medline, Google Scholar
20. Roy CW, Sherrington CS: On the regulation of the blood supply of the brain. J Physiol (Lond) 1980; 11:85–108Crossref, Google Scholar
21. Nilsson B, Rehncrona S, Siesjo BK: Coupling of cerebral metabolism and blood flow in epileptic seizures, hypoxia, and hypoglycaemia. Ciba Found Symp 1978; 56:199–218Medline, Google Scholar
22. Des Rosiers MH, Kennedy C, Patlak CS, et al: Relationship between local cerebral blood flow and glucose utilization in the rat. Neurology 1974; 24:389Google Scholar
23. Raichle ME, Grubb R, Gado MH, et al: Correlation between regional cerebral blood flow and oxidative mechanisms. Arch Neurol 1976; 33:523–526Crossref, Medline, Google Scholar
24. Ingvar DH, Lassen NA: Activity distribution in the cerebral cortex in organic dementia as revealed by measurements of regional cerebral blood flow, in Brain Function in Old Age, edited by Hochmeister F, Muller C. New York, Springer-Verlag, 1979, pp 268–277Google Scholar
25. Schieve JF, Wilson WP: The influence of age, anaesthesia and cerebral arteriosclerosis on cerebral vascular activity to CO2. Am J Med 1953; 15:171–174Crossref, Medline, Google Scholar
26. Porrino LJ: Functional effects of cocaine depend upon route of administration. Psychopharmacology 1993; 112:343–351Crossref, Medline, Google Scholar
27. London ED, Cascella NG, Wong DF, et al: Cocaine induced reduction of glucose utilization in human brain. Arch Gen Psychiatry 1990; 47:567–574Crossref, Medline, Google Scholar
28. Pearlson GD, Jeffery PJ, Harris GJ, et al: Correlation of acute cocaine-induced changes in local cerebral blood flow with subjective effects. Am J Psychiatry 1993; 150:495–497Crossref, Medline, Google Scholar
29. Volkow ND, Hitzemann R, Wang GJ, et al: Long-term frontal metabolic changes in cocaine abusers. Synapse 1992; 11:184–190Crossref, Medline, Google Scholar
30. Stapleton JM, Morgan MJ, Phillips RL, et al: Cerebral glucose utilization in polysubstance abuse. Neuropsychopharmacology 1995; 13:21–31Crossref, Medline, Google Scholar
31. Mena I, Miller B, Garrett K, et al: Neurospect in cocaine abuse (abstract). Eur J Nucl Med 1990; 16:5S137Google Scholar
32. Tumeh SS, Nagel SJ, English RJ, et al: Use of SPECT perfusion brain scintigraphy to investigate effects of cocaine on the brain, in Physiopathology of Illicit Drugs: Cannabis, Cocaine Opiates, edited by Nahas GG, Latour G. Oxford, UK, Pergamon Press, 1991, pp 143–151Google Scholar
33. Strickland TL, Mena I, Villanueva-Meyer J, et al: Cerebral perfusion and neuropsychological consequences of chronic cocaine use. J Neuropsychiatry Clin Neurosci 1993; 5:419–427Link, Google Scholar
34. Sloan MA, Mattioni TA: Concurrent myocardial and cerebral infarctions after intranasal cocaine use. Stroke 1992; 23:427–430Crossref, Medline, Google Scholar
35. Klonoff DC, Andrews BT, Obana WG: Stroke associated with cocaine use. Arch Neurol 1989; 46:989–993Crossref, Medline, Google Scholar
36. Levine SR, Brust JCM, Futrell N, et al: Cerebrovascular complications of the use of the “crack” form of alkaloidal cocaine. N Engl J Med 1990; 323:699–704Crossref, Medline, Google Scholar
37. Levine SR, Brust JCM, Futrell N, et al: A comparative study of the cerebrovascular complications of cocaine: alkoloidal versus hydrochloride. A review. Neurology 1991; 41:1173–1177Crossref, Medline, Google Scholar
38. Daras M, Tuchman AJ, Marks S: Central nervous system infarction related to cocaine abuse. Stroke 1991; 22:1320–1325Crossref, Medline, Google Scholar
39. Powers RH, Madden JA: Vasoconstrictive effects of cocaine metabolites, and structural analogs on cat cerebral arteries (abstract). FASEB J 1990; 4:A1095Google Scholar
40. Isner JM, Chokshi SK: Cocaine and vasospasm. N Engl J Med 1989; 321:1604–1606Crossref, Medline, Google Scholar
41. Rumbaugh C, Bergeron T, Scanlan R, et al: Cerebral vascular changes secondary to amphetamine abuse in the experimental animal. Radiology 1971; 101:345–351Crossref, Medline, Google Scholar
42. Hardebo JE, Edvinson L, Owman C, et al: Potentiation and antagonism of serotonin effects on intracranial and extracranial vessels: possible implications in migraine. Neurology 1978; 28:64–70Crossref, Medline, Google Scholar
43. Mody CK, Miller BL, McIntyre HB, et al: Neurologic complications of cocaine abuse. Neurology 1988; 38:1189–1193Crossref, Medline, Google Scholar
44. Rowley HA, Lowenstein DH, Rowbotham MC, et al: Thalamomesencephalic strokes after cocaine abuse. Neurology 1989; 39:428–430Crossref, Medline, Google Scholar
45. Levine SR, Welch KMA: Cocaine and stroke. Stroke 1988; 19:779–783Crossref, Medline, Google Scholar
46. Pascual-Leone A, Dhuna A, Anderson DC: Cerebral atrophy in habitual cocaine abusers: a planimetric CT study. Neurology 1991; 41:34–38Crossref, Medline, Google Scholar
47. Langendorf FG, Anderson DC, Tupper DE, et al: Brain atrophy and chronic cocaine abuse: background and work in progress. Rockville, MD, U.S. Dept. of Health and Human Services, National Institutes of Health. National Institute on Drug Abuse Research Monograph Series: Neurotoxicity and Neuropathology Associated with Cocaine Abuse, 1996Google Scholar
48. Cascella NG, Pearlson G, Wong DF, et al: Effects of substance abuse on ventricular and sulcal measures assessed by computerized tomography. Br J Psychiatry 1991; 159:217–221Crossref, Medline, Google Scholar
49. Liu X, Phillips RL, Resnick SM, et al: Magnetic resonance imaging reveals no ventriculomegaly in polydrug abusers. Acta Neurol Scand 1995; 92:83–90Crossref, Medline, Google Scholar
50. Berry J, Van Gorp WG, Herzberg DS, et al: Neuropsychological deficits in abstinent cocaine abusers: preliminary findings after two weeks of abstinence. Drug Alcohol Depend 1993; 32:231–237Crossref, Medline, Google Scholar
51. Meek PS, Clark HW, Solana VL: Neurocognitive impairment: the unrecognized component of dual diagnosis in substance abuse treatment. J Psychoactive Drugs 1989; 21:153–160Crossref, Medline, Google Scholar
52. Manschreck TC, Schneyer ML, Weisstein CC, et al: Freebase cocaine and memory. Compr Psychiatry 1990; 31:369–375Crossref, Medline, Google Scholar
53. Ardila A, Rosselli M, Strumwasser S: Neuropsychological deficits in chronic cocaine abusers. Int J Neurosci 1991; 57:73–79Crossref, Medline, Google Scholar
54. Hoff AL, Riordan H, Alpert R, et al: Cognitive function in chronic cocaine abusers (abstract). J Clin Exp Neuropsychol 1991; 13:60Google Scholar
55. Mittenburg W, Motta S: Effects of chronic cocaine abuse on memory and learning. Arch Clin Neuropsychol 1993; 8:477–483Crossref, Medline, Google Scholar
56. Weber DA, Klieger P, Volkow ND, et al: SPECT regional cerebral blood flow (rCBF) studies in crack users and control participants. J Nucl Med 1990; 31:876–877Google Scholar
57. Stuss DT, Benson DF: The Frontal Lobes. New York, Raven, 1986Google Scholar
58. Ohnishi T, Hoshi H, Nagamachi S, et al: High resolution SPECT to assess hippocampal perfusion in neuropsychiatric diseases. J Nucl Med 1995; 36:1163–1169Medline, Google Scholar
59. Lezak MD: Neuropsychological Assessment, 3rd edition. New York, Oxford University Press, 1995Google Scholar
60. Wide RJ, Bernardi S, Frackowiak R, et al: Serial observation on the pathophysiology of acute stroke: the transition from ischemia to infarction as reflected in regional oxygen extraction. Brain 1983; 106:197–222Crossref, Medline, Google Scholar
61. Gawin FH, Kleber HD: Abstinence symptomatology and psychiatric diagnoses in cocaine abusers: clinical observations. Arch Gen Psychiatry 1986; 43:107–113Crossref, Medline, Google Scholar
62. Stein DJ, Hollander E, Cohen L: Neuropsychiatric impairment in impulsive personality disorders. Psychiatry Res 1993; 48:257–266Crossref, Medline, Google Scholar
63. Bauer LO, O'Connor S, Hesselbrock VM: Frontal P300 decrements in antisocial personality disorder. Alcohol Clin Exp Res 1994: 18:1300–1305Google Scholar
64. Bechara A, Damasio AR, Damasio H, et al: Insensitivity to future consequences following damage to human prefrontal cortex. Cognition 1994; 50:7–15Crossref, Medline, Google Scholar
65. Rolls ET, Baylis LL: Gustatory, olfactory, and visual convergence within the primate orbitofrontal cortex. J Neurosci 1994; 14:5437–5452Crossref, Medline, Google Scholar
66. Rolls ET, Burton MJ, Mora F: Neurophysiological analysis of brain-stimulation reward in the monkey. Brain Research 1980; 194:339–357Crossref, Medline, Google Scholar
67. Mora F, Avrith DB, Rolls ET: An electrophysiological and behavioral study of self-stimulation in the orbitofrontal cortex of the rhesus monkey. Brain Res Bull 1980; 5:111–115Crossref, Medline, Google Scholar
68. Grant S, London ED, Newlin DB, et al: Activation of memory circuits during cue-elicited cocaine craving. Proc Natl Acad Sci USA 1996; 93:12040–12045Crossref, Medline, Google Scholar
69. Baxter LRJ, Phelps ME, Mazziotta JC, et al: Local cerebral glucose metabolic rates in obsessive-compulsive disorder: a comparison with rates in unipolar depression and in normal controls. Arch Gen Psychiatry 1987; 44:211–218Crossref, Medline, Google Scholar
70. Benkelfat C, Nordahl TE, Semple WE, et al: Local cerebral glucose metabolic rates in obsessive-compulsive disorder. Arch Gen Psychiatry 1990; 47:840–848Crossref, Medline, Google Scholar
71. Dworkin SI, Smith JE: Cortical regulation of self-administration in Neurobiology of Drug and Alcohol Addiction, edited by Kalivas PW, Samson HA. New York, New York Academy of Sciences, 1992, pp 274–281Google Scholar
72. Paus T, Petrides M, Evans A, et al: Role of human anterior cingulate cortex in the control of oculomotor, manual, and speech responses: a positron emission tomography study. J Neurophysiol 1993; 70:453–469Crossref, Medline, Google Scholar
73. Johanson CE, Fischman MW: The pharmacology of cocaine related to its abuse. Pharmacol Rev 1989; 41:3–52Medline, Google Scholar
74. Wang GJ, Volkow ND, Roque CT, et al: Functional importance of ventricular enlargement and cortical atrophy in healthy participants and alcoholics as assessed with PET, MR imaging, and neuropsychological testing. Radiology 1993; 186:59–65Crossref, Medline, Google Scholar
75. Bolla KI, Schwartz BS, Stewart W, et al: A comparison of neurobehavioral function in workers exposed to a mixture of organic and inorganic lead and in workers exposed to solvents. Am J Ind Med 1995; 27:231–246Crossref, Medline, Google Scholar
76. Bleecker ML, Bolla I, Agnew J, et al: Dose-related subclinical neurobehavioral effects of chronic exposure to low levels of organic solvents. Am J Ind Med 1991; 19:715–728Crossref, Medline, Google Scholar
77. Cadet JL, Landenheim B, Hiroshi H, et al: Superoxide radicals mediate the biochemical effects of methylenedioxymethamphetamine (MDMA): Evidence from using CuZn-superoxide dismutase transgenic mice. Synapse 1995; 21:169–176Crossref, Medline, Google Scholar
78. Dolan RJ, Friston KJ: Positron emission tomography in psychiatric and neuropsychiatric disorders. Semin Neurol 1989; 9:330–337Crossref, Medline, Google Scholar
79. Petersen SE, Fox BT, Posner MI, et al: Positron emission tomographic studies of the cortical anatomy of single-word processing. Nature 1988; 331:585–589Crossref, Medline, Google Scholar
80. Grasby PM, Friston KJ, Bench CJ, et al: The effect of apomorphine and buspirone on regional cerebral blood flow during the performance of a cognitive task: measuring neuromodulatory effects of psychotropic drugs in man. Eur J Neurosci 1992; 4:1203–1212Crossref, Medline, Google Scholar
81. Squire LR, Ojemann JG, Miezin FM, et al: Activation of the hippocampus in normal humans: a functional anatomical study of memory. Proc Natl Acad Sci USA 1992; 89:1837–1841Crossref, Medline, Google Scholar
82. Weinberger DR, Berman KF, Zec RF: Physiologic dysfunction of the dorsolateral prefrontal cortex in schizophrenia: regional cerebral blood flow evidence. Arch Gen Psychiatry 1986; 43:114–124Crossref, Medline, Google Scholar

