
Blood–Brain Barrier Integrity in Alzheimer's Disease Patients and Elderly Control Subjects
Abstract
A defective blood–brain barrier (BBB) has been postulated to be present in Alzheimer's disease (AD), which would allow circulating β-amyloid peptide to enter the brain. The authors tested this hypothesis by studying BBB function in 14 individuals with probable AD and 9 elderly control subjects. A computed tomographic method was used to measure blood-to-brain transport (K1), tissue-to-blood efflux (k2), tissue plasma space (Vp), and tissue extracellular space (Ve) of meglumine iothalamate. Repeated-measures analysis of variance indicated no significant differences between the groups for any of the measures. The authors conclude that there is no generalized abnormality of the blood–brain barrier in AD.
Despite enormous strides in the past few years, the etiology and pathogenesis of Alzheimer's disease (AD) are still not well understood. It is now established that the apolipoprotein E type 4 allele accounts for increased genetic susceptibility to AD1 and that there are three identified genes on three different chromosomes that lead to familial AD.2–5 The roles of zinc,6 immune-mediated mechanisms,7 decreased glucose metabolism,8 a possible amyloid prion,9 and other contributing factors remain unclear.10
The neuropathological hallmarks of Alzheimer's disease are increased extracellular amyloid plaques, both perivascularly and intraparenchymally, and the concomitant presence of intraneuronal neurofibrillary tangles. These are seen in the hippocampus in the early stages of AD and are believed to underlie the memory deficits that are the harbingers of this disease. Neuropathological changes involve all other areas of the cortex in later stages of the disease. The major component of the cerebral amyloid deposits is the βA4 protein,11 which consists of 40–42 amino acids and is derived from amyloid precursor protein (APP), a membrane-associated protein.12 The origin of the βA4 protein in AD has not been established: both hematogenous and intracerebral origins have been proposed.11,12
Some investigators have postulated that the blood–brain barrier (BBB) is defective in AD,13 which would allow βA4 amyloid protein to enter the brain from the systemic circulation. Most evidence for an abnormal BBB has been indirect, such as increased CSF protein levels, immunocytochemical staining of plasma proteins, subtle changes in brain capillary structure, and a geographic association between brain capillaries and amyloid plaques. Two previous studies using positron emission tomography (PET) in Alzheimer's patients reported that BBB function was normal.14,15 In this study, we reevaluated the possibility of abnormal BBB function in AD subjects by using a different method to quantitatively measure BBB function in living subjects. We compared BBB function in AD subjects with that in elderly control subjects.
METHODS
Selection of Subjects
AD subjects and control subjects were residents of the Presbyterian Homes in Evanston, IL, a continuing-care retirement community that included a 99-bed Alzheimer's unit. Medical and cognitive screening was performed regularly, and a unitary medical record was maintained. The control group was matched in age and education and subjects were independent, with no cognitive impairment. The 14 AD subjects were from a “mildly to moderately” cognitively impaired group with a diagnosis of probable AD.16 We selected mildly to moderately impaired individuals who were still relatively functional and independent for two reasons: 1) we wanted subjects to cooperate with the CT procedure without psychopharmacological intervention, and 2) we hypothesized that subjects in earlier phases of the disease process were more likely to show blood–brain barrier changes than those in end stages. AD and control subjects were screened medically, neurologically, and cognitively prior to the study and were excluded if they demonstrated evidence of neurological disease other than AD. Most AD and control subjects were on medication for hypertension, adult-onset diabetes, arthritis, depression, cardiac disease, or hypothyroidism; their medical conditions were stable. Subjects taking chronic medication were not excluded, nor was there a washout period prior to the study. Blood counts, electrolytes, and liver and thyroid function tests were normal. Cognitive impairment was measured by the CAST (Cognitive Assessment and Screening Test), which is a combination of the Mini-Mental State Examination17 and the Boston Naming Test18 and is routinely used for following patients at the Presbyterian Homes. A perfect CAST score was 88; normal was 82–88; mildly impaired was 70–81; moderately impaired was 30–69; and severely impaired was <30.
Performance of CT Scans
This project was approved by the Institutional Review Board of Evanston Hospital Corporation. Informed consent was obtained from each subject or his or her legal guardian. Each subject was positioned in a General Electric (Milwaukee, WI) CT MAX 640 scanner so that CT slices were obtained at a level through the hippocampus. The methods have been described19,20 and are presented briefly here. Meglumine iothalamate (2 ml/kg, Conray-60™ Mallinckrodt, St. Louis, MO) was infused over 5 minutes. Scans were performed with 120 kV beam voltage, 55 ma beam current, and 4.8 seconds of continuous scanning per image. The volume element resolution (voxel) size was 0.8×0.8×2.0 mm. Twenty CT scans were made over 60 minutes. After the study, a complete series of CT scans was taken and evaluated for incidental pathology.
The CT scan series was transferred to an image processing computer. Postinfusion scans were registered to the background scan. The time course of the plasma concentration of meglumine iothalamate [Cp(t)] was obtained from the CT scan series. The amount of meglumine iothalamate in tissue [Am(t)], was obtained by subtracting each voxel of the background scan from the corresponding voxel of each CT scan in the timed series.21 The experimentally obtained data, Am(t) and Cp(t), were fit to the following equation with nonlinear least-squares methods to obtain K1 (blood-to-tissue transfer constant; ml·g–1·min–1, k2 (blood-to-tissue efflux constant; min–1), and Vp (tissue plasma space; ml/g)19,20: 
The range of k2 was fixed from 0 to 0.1, in increments of 0.01 min–1. The terms K1 and k2 were related by the expression 
where λ was the equilibrium partition coefficient, with units of ml/g. We used λ as an estimate of the size of the tissue extracellular space; that is, λ≈Ve, where Ve was the fractional volume of the extracellular space, with units of ml/g.
Physiological images of K1, k2, Vp, and Ve were created and analyzed to obtain regional values of each parameter for each brain region by outlining a region of interest and obtaining the mean±SD of the values for all voxels within that region. Values were obtained from the following regions of both hemispheres: frontal lobe, basal ganglia, temporal lobe, hippocampus, and occipital lobe. Regions of interest included both gray and white matter and were 150–400 voxels (96–256 mm2) in area.
Statistical Analysis
Demographic characteristics were compared with Student's t-test. The regional measures of blood–brain barrier function in the two groups were compared with repeated-measures analysis of variance (MANOVA). All significance tests were two-tailed.
RESULTS
There were 14 AD subjects (13F:1M) and 9 control subjects (8F:1M). The AD subjects were slightly older than control subjects (87.3±3.8 vs. 83.4±3.1 years; t=2.54, df=21, P=0.02). The AD subjects had significantly lower CAST scores than the control subjects (57.0±17.5 vs. 84.0±3.0; t=–4.56, df=20, P<0.01). Eleven AD subjects scored in the 40–70 range; the other 3 were severely impaired (<30) but were functioning at the moderately impaired level. All control subjects scored in the 80–88 range and were fully functional. CT scans from the AD subjects showed moderate to severe atrophy (Figure 1). Two AD subjects had small old infarctions on their CT scans, and one had calcification of the basal ganglia. Eliminating these subjects from the data analysis did not alter the results. The CT scans of the control subjects did not demonstrate any significant pathology.
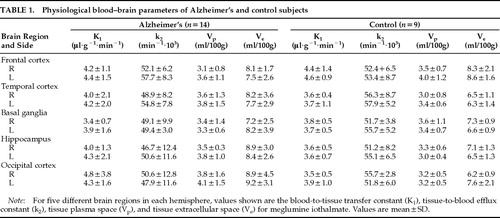
Reconstructed images of the rate of blood-to-tissue transport (K1) of meglumine iothalamate are shown in Figure 2. Table 1 presents the values from each of the 10 brain regions that were studied.
For each of the physiological values (K1, k2, Vp, and Ve) we used repeated-measures analysis of variance to examine the physiological values collected from five different regions of each hemisphere in the two groups. Initial examination of the data indicated that the distributional assumptions for repeated measures of variance were satisfied. The data for K1, k2, Vp, and Ve were then analyzed independently in four separate MANOVAs. The analysis indicated no significant main effect for diagnosis and no significant interactions between diagnosis and region or hemisphere. Planned contrast of the hippocampal region, an area of well-established pathology in AD, with the other regions indicated no significant difference between the groups. Thus, no difference between AD and control subjects was found for the principal blood–brain barrier measure, K1. We did not find any significant differences between the two groups for k2 or for Vp. A nearly significant main effect for diagnosis was obtained for Ve (F=3.96, df=1,20, P=0.06), a measure of tissue extracellular space. As shown in Table 1, the AD subjects had higher values on this measure in 7/10 brain regions. This finding was consistent with the presence of greater atrophy among the AD subjects, as observed on CT scan, rather than loss of blood–brain barrier integrity.
DISCUSSION
This study of quantitative transcapillary transport of meglumine iothalamate (Conray-60™), a water-soluble iodinated contrast agent, did not show any difference in the rate of blood-to-brain transport in living AD subjects compared with elderly control subjects. This study used a computed tomographic method with a volume element resolution (voxel size) of 0.8×0.8×2.0 mm and confirmed findings from two previous permeability studies in AD subjects that were done with PET. The PET studies had a much larger voxel size (1.7 cm) and used 82Rb and 68Ga-EDTA (ethylenediaminetetraacetic acid).14,15 These three studies, using different markers of capillary permeability and performed with different methodologies, have demonstrated that there is no generalized increase in BBB permeability in living AD subjects. These observations do not support the concept that an abnormal BBB is a step in the pathogenesis of AD.
The premise that the BBB in AD is abnormal rests on four different types of evidence:
| 1. | Histological abnormalities of brain capillaries. | ||||
| 2. | Immunocytochemical demonstration of extravasated plasma proteins in postmortem brain tissue. | ||||
| 3. | Abnormally high levels of albumin (and/or other proteins) in the cerebrospinal fluid (CSF). | ||||
| 4. | A potential relationship between the spatial distribution of brain capillaries and βA4 protein. | ||||
First, there are many reports of altered microvasculature in AD. These include ultrastructural changes in capillaries22–25 as well as changes in capillary distribution and density.24,26,27 This evidence creates an apparent contradiction between the functional PET and CT studies. Perlmutter22 suggested that BBB disturbances might be in the form of “microleaks” rather than a “severe loss of BBB integrity.” This statement assumes that the functional imaging techniques are not sensitive enough to detect very small changes in BBB function. It is worth noting that none of the ultrastructural studies in AD have shown capillary fenestrations or interendothelial gaps, which are classically associated with increased capillary permeability in other intracranial diseases. Perhaps the truth lies somewhere in the middle: perhaps βA4 protein processing injures brain capillaries,27,28 but not sufficiently to interrupt functional integrity.
Second, the immunocytochemical evidence is based on the demonstration of extravasated serum proteins in histological sections of brain from human AD subjects.29,30 Although a variety of techniques were used in these studies, all but one demonstrated increased albumin and/or other proteins in postmortem brain. Rozemuller et al.31 concluded that this method was unreliable in postmortem specimens, stating that there are “no convincing arguments for BBB dysfunction in Alzheimer's dementia.”
The third type of evidence of possible abnormal BBB function in AD is increased levels of plasma proteins in CSF. Although occasional studies of CSF proteins have questioned a relationship between AD and the blood–CSF barrier,32 others treated increased serum proteins in CSF as evidence of increased BBB permeability in AD.33 The primary source of protein in CSF is from choroid plexus and not the brain.34 Alterations in CSF protein composition may, therefore, reflect pathological changes in the choroid plexus and have little relationship to parenchymal changes in the brain.
The fourth type of support for an abnormal BBB in AD is from histological observations of brain capillaries and their relationship to amyloid deposits. Early studies suggested that there may be a spatial relationship between blood vessels and βA4 deposits or plaques,35,36 which subsequent studies have been unable to confirm.37,38 It is now becoming clear that the normal, as well as abnormal, processing of βA4 protein occurs in many cells within the nervous system39–41 and that involvement of the microvasculature is part of the disease process rather than the cause.
Although the etiology and pathogenesis of AD are not clearly established, it may not be necessary to invoke an abnormal BBB as part of the pathogenic mechanism. If APP is a normal constituent of cells, it is likely that abnormal proteolytic processing accounts for the production of βA4 protein, which may occur throughout the body.40,41 βA4 protein in the brain could come from both neural and hematogenous sources. Maness et al.42 demonstrated that βA4 protein crosses the normal murine blood–brain barrier and that βA4 protein uptake by the brain is nonsaturable, although Zlokovic et al.43 suggested that uptake is saturable. Zlokovic et al. also demonstrated that brain uptake of βA4 protein is increased 100-fold when it is complexed with apolipoprotein J.44 These results imply an active uptake process for βA4 across the BBB rather than a passive one. Although the relative importance of the roles of βA4 protein and the apolipoproteins is still controversial,45,46 it seems most likely that the blood–brain barrier is not part of the pathogenesis of this disease. Furthermore, our functional study suggests that compared with the severe degree of injury suffered by neural cells, the blood–brain barrier remains normal in Alzheimer's disease.
ACKNOWLEDGMENTS
The authors thank Monte Levinson, M.D., Medical Director, and Lydia Torrese, M.S.N., R.N., of the Presbyterian Homes, for their assistance in identifying the subjects and facilitating participation, and Cathleen Allen, M.S., R.T., for her assistance in performing the CT scans. This work was supported by a grant from the Washington Square Health Foundation, by National Institutes of Health Grant S10-RR03321, and by a donation from the Searle Family Center for Neurological Disorders of Northwestern University Medical School. These findings were a presented at the Society for Neuroscience 22nd annual meeting, November 1992.
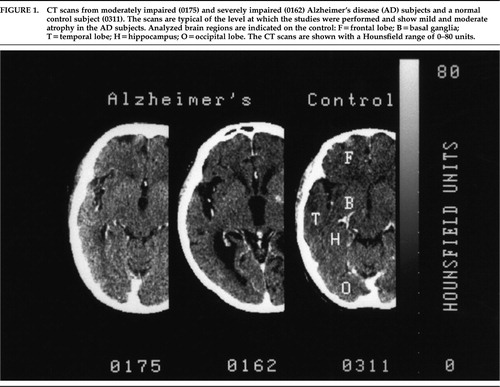
FIGURE 1. CT scans from moderately impaired (0175) and severely impaired (0162) Alzheimer's disease (AD) subjects and a normal control subject (0311). The scans are typical of the level at which the studies were performed and show mild and moderate atrophy in the AD subjects. Analyzed brain regions are indicated on the control: F=frontal lobe; B=basal ganglia; T=temporal lobe; H=hippocampus; O=occipital lobe. The CT scans are shown with a Hounsfield range of 0–80 units.
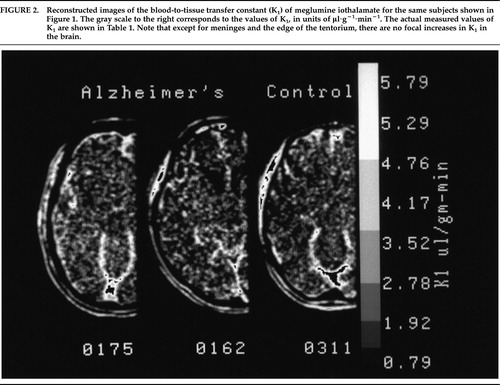
FIGURE 2. Reconstructed images of the blood-to-tissue transfer constant (K1) of meglumine iothalamate for the same subjects shown in Figure 1. The gray scale to the right corresponds to the values of K1, in units of μl·g–1·min–1. The actual measured values of K1 are shown in Table 1. Note that except for meninges and the edge of the tentorium, there are no focal increases in K1 in the brain.
 |
1. Strittmatter WJ, Roses AD: Apolipoprotein E and Alzheimer disease. Proc Natl Acad Sci USA 1995; 92:4725–4727Crossref, Medline, Google Scholar
2. St. George-Hyslop PH, Tangi RE, Polinsky RJ, et al: The genetic defect causing familial Alzheimer's disease maps on chromosome 21. Science 1987; 235:885–890Crossref, Medline, Google Scholar
3. Rogaev EI, Sherrington R, Rogaeva EA, et al: Familial Alzheimer's disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer's disease type 3 gene. Nature 1995; 376:775–778Crossref, Medline, Google Scholar
4. Schellenberg GD, Bird TD, Wijsman EM, et al: Genetic linkage evidence for a familial Alzheimer's disease locus on chromosome 14. Science 1992; 258:668–671Crossref, Medline, Google Scholar
5. Mullan M, Houlden H, Windelspecht M, et al: A locus for familial early onset Alzheimer's disease on the long arm of chromosome 14, proximal to the alpha 1 antichymotrypsin gene. Nat Genet 1992; 2:340–342Crossref, Medline, Google Scholar
6. Bush AI, Pettingell WH, Multhaup G, et al: Rapid induction of Alzheimer A beta amyloid formation by zinc. Science 1995; 265:1464–1467Crossref, Google Scholar
7. Kalaria RN: The immunopathology of Alzheimer's disease and some related disorders. Brain Pathol 1993; 3:333–347Crossref, Medline, Google Scholar
8. Meier-Ruge W, Bertoni-Freddari C, Iwangoff P: Changes in brain glucose metabolism as a key to the pathogenesis of Alzheimer's disease. Gerontol 1994; 40:246–252Crossref, Medline, Google Scholar
9. Price DL, Borchelt DR, Sisodia SS: Alzheimer disease and the prion disorders β-amyloid protein and prion protein amyloidoses. Proc Natl Acad Sci USA 1993; 90:6381–6384Crossref, Medline, Google Scholar
10. Itzhaki RF: Possible factors in the etiology of Alzheimer's disease. Mol Neurobiol 1994; 9:1–13Crossref, Medline, Google Scholar
11. Wong CW, Quaranta V, Glenner CG: Neuritic plaques and cerebrovascular amyloid in Alzheimer's disease are antigenically related. Proc Natl Acad Sci USA 1985; 82:8729–8732 Crossref, Medline, Google Scholar
12. Kang J, Lemaire HG, Unterbeck A, et al: The precursor of Alzheimer's disease amyloid A4 protein resembles a cell-surface receptor. Nature 1987; 325:733–736Crossref, Medline, Google Scholar
13. Kalaria RN: The blood–brain barrier and cerebral microcirculation in Alzheimer disease. Cerebrovascular Brain Metabolism Review 1992; 4:226–260Medline, Google Scholar
14. Freidland RP, Yano Y, Budinger TF, et al: Quantitative evaluation of blood–brain barrier integrity in Alzheimer type dementia: positron emission tomographic studies with rubidium-82. Eur Neurol 1983; 22(suppl 2):15–20Google Scholar
15. Schlageter NL, Carson RE, Rapoport SI: Examination of blood–brain barrier permeability in dementia of the Alzheimer type with [68Ga]EDTA and positron emission tomography. J Cereb Blood Flow Metab 1987; 7:1–8Crossref, Medline, Google Scholar
16. McKhann G, Drachman D, Folstein M, et al: Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA work group under auspices of Department of Health and Human Services Task Force on Alzheimer's disease. Neurology 1984; 34:939–944Crossref, Medline, Google Scholar
17. Folstein M, Folstein S, McHugh PR: “Mini-Mental State”: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975; 12:189–198Crossref, Medline, Google Scholar
18. Corbel H, Domoto K, Nobel K, et al: Interdisciplinary approach to the development of a cognitive screening test for the institutionalized elderly. Presented to the Gerontological Society of America, San Francisco, CA, 1983Google Scholar
19. Groothuis DR, Vriesendorp FJ, Lapin GD, et al: Quantitative measurement of transcapillary transport of iodinated compounds in canine brain tumors with computed tomography. J Cereb Blood Flow Metab 1991; 11:939–948Crossref, Medline, Google Scholar
20. Groothuis DR, Vriesendorp FJ, Kupfer B, et al: Quantitative measurements of capillary transport in human brain tumors by computed tomography. Ann Neurol 1991; 30:581–588Crossref, Medline, Google Scholar
21. Lapin DG, Allen CA, Groothuis DR: Noninvasive measurement of arterial blood plasma concentration of iodinated contrast agents from CT scans of human brain. J Comput Assist Tomogr 1994; 18:363–369Crossref, Medline, Google Scholar
22. Perlmutter LS: Microvascular pathology and vascular basement membrane components in Alzheimer's disease. Mol Neurobiol 1994; 9:33–40Crossref, Medline, Google Scholar
23. Vinters HV, Secor DL, Read SL, et al: Microvasculature in brain biopsy specimens from patients with Alzheimer's disease: an immunohistochemical and ultrastructural study. Ultrastruct Pathol 1994; 18:333–348Crossref, Medline, Google Scholar
24. Buee L, Hof PR, Bouras C, et al: Pathological alterations of the cerebral microvasculature in Alzheimer's disease and related dementing disorders. Acta Neuropathol 1994; 87:469–480Crossref, Medline, Google Scholar
25. Claudio L: Ultrastructural features of the blood–brain barrier in biopsy tissue from Alzheimer's disease patients. Acta Neuropathol 1996; 91:6–14Crossref, Medline, Google Scholar
26. Kalaria RN, Hedera P: Differential degeneration of the cerebral microvasculature in Alzheimer's disease. NeuroReport 1995; 6:477–480Crossref, Medline, Google Scholar
27. Perlmutter LS, Myers MA, Barron E: Vascular basement membrane components and the lesions of Alzheimer's disease: light and electron microscopic analyses. Microsc Res Tech 1994; 28:204–215Crossref, Medline, Google Scholar
28. Roher AE, Lowenson JD, Clarke S, et al: Beta-amyloid-(1-42) is a major component of cerebrovascular amyloid deposits: implications for the pathology of Alzheimer disease. Proc Natl Acad Sci USA 1993; 90:10836–10840Crossref, Medline, Google Scholar
29. Behrouz N, Defossez A, Delacourte A, et al: The immunohistochemical evidence of amyloid diffuse deposits as a pathological hallmark in Alzheimer's disease. J Gerontol Biol Sci 1991; 46:B209–B212Google Scholar
30. Eikelenboom P, Stam FC: Neuroimmunological mechanisms in cerebral amyloid deposition in Alzheimer's disease, in Frontiers of Alzheimer Research, edited by Ishii T. Amsterdam, Elsevier, 1991, pp 259–271Google Scholar
31. Rozemuller JM, Eikelenboom P, Kamphorst W, et al: Lack of evidence for dysfunction of the blood–brain barrier in Alzheimer's disease: an immunohistochemical study. Neurobiol Aging 1988; 9:383–391Crossref, Medline, Google Scholar
32. Frolich L, Kornhuber J, Ihl R, et al: Integrity of the blood–CSF barrier in dementia of Alzheimer type: CSF/serum ratios of albumin and IgG. Eur Arch Psychiatry Clin Neurosci 1991; 240:363–366Crossref, Medline, Google Scholar
33. Mattila KM, Pirttila T, Blennow K, et al: Altered blood-brain–barrier function in Alzheimer's disease? Acta Neurol Scand 1994; 89:192–198Google Scholar
34. Wood JH: Physiology, pharmacology, and dynamics of cerebrospinal fluid, in Neurobiology of Cerebrospinal Fluid, edited by Wood JH. New York, Plenum, 1980, pp 1–16Google Scholar
35. Arai H, Sagi N, Noguchi I, et al: An immunohistochemical study of β-protein in Alzheimer-type dementia brains. J Neurol 1989; 236:214–217Crossref, Medline, Google Scholar
36. Miyakawa T, Shomoji A, Kuramoto R, et al: The relationship between senile plaques and cerebral blood vessels in Alzheimer's disease and senile dementia. Virchows Archiv Path 1982; 40:121–129Crossref, Google Scholar
37. Lippa CF, Hamos JE, Smith TW, et al: Vascular amyloid deposition in Alzheimer's disease: neither necessary nor sufficient for the local formation of plaques or tangles. Arch Neurol 1993; 50:1088–1092Crossref, Medline, Google Scholar
38. Luthert PJ, Williams JA: A quantitative study of the coincidence of blood vessels and A4 protein deposits in Alzheimer's disease. Neurosci Lett 1991; 126:110–112Crossref, Medline, Google Scholar
39. Wisniewski HM, Frackowiak J, Zoltowska AS, et al: Vascular β-amyloid in Alzheimer's disease angiopathy is produced by proliferating and degenerating smooth muscle cells. Int J Exp Clin Invest 1994; 1:8–16Google Scholar
40. Gandy S, Greengard P: Processing of Alzheimer A β-amyloid precursor protein: cell biology, regulation, and role in Alzheimer disease. Int Rev Neurobiol 1994; 36:29–50Crossref, Medline, Google Scholar
41. Selkoe DJ: Cell biology of the amyloid β-protein precursor and the mechanism of Alzheimer's disease. Annu Rev Cell Biol 1994; 10:373–403Crossref, Medline, Google Scholar
42. Maness LM, Banks WA, Podlisny MB, et al: Passage of human amyloid beta-protein 1-40 across the murine blood–brain barrier. Life Sci 1994; 55:1643–1650Crossref, Medline, Google Scholar
43. Zlokovic BV, Ghiso J, Mackic JB, et al: Blood–brain barrier transport of circulating Alzheimer's amyloid beta. Biochem Biophys Res Comm 1993; 197:1034–1040Crossref, Medline, Google Scholar
44. Zlokovic BV, Martel CL, Mackic JB, et al: Brain uptake of circulating apolipoproteins J and E complexed to Alzheimer's amyloid beta. Biochem Biophys Res Commun 1994; 205:1431–1437Crossref, Medline, Google Scholar
45. Roses AD: Apolipoprotein E affects the rate of Alzheimer disease expression: β-amyloid burden is a secondary consequence dependent upon APOE genotype and duration of disease. J Neuropathol Exp Neurol 1994; 53:429–437Crossref, Medline, Google Scholar
46. Selkoe DJ: Alzheimer's disease: a central role for amyloid. J Neuropathol Exp Neurol 1994; 53:438–447Crossref, Medline, Google Scholar

