
The Psychiatric Presentation of Mitochondrial Disorders in Adults
Abstract
Mitochondrial disorders are clinical syndromes produced by a primary impairment of mitochondrial functioning. Mitochondria are intracellular organelles that play a critical role in cellular energy metabolism through the Krebs cycle and respiratory chain. They contain their own 16.6 Kb circular, double-stranded genome, which is maternally inherited and which encodes for 13 subunits of the respiratory chain, in addition to 2 rRNAs and 22 tRNAs.1 Also, many of the polypeptides that form the respiratory chain complexes are encoded by nuclear DNA and are transported into the mitochondria from the cytosol. As a result, mutations in either nuclear DNA or mitochondrial DNA can cause mitochondrial disorders.2,3
Mitochondrial disorders are much more common than previously appreciated, with an estimated prevalence of 9.2 per 100, 000 adults for mutations in mitochondrial DNA alone.4 These disorders may present at any age and typically involve organs that are highly energy-dependent, such as muscle and brain. Common clinical features of mitochondrial disorder include fatigue, ptosis, ophthalmoplegia, optic atrophy, hearing loss, seizures, migraine, stroke-like episodes, ataxia, cardiomyopathy, diabetes mellitus, and proximal myopathy.2,5,6
Since the description of the first mitochondrial gene mutation in 1988,7 its medical and neurological complications have come to be well characterized. By contrast, the literature on patients presenting initially with psychiatric symptoms is limited to sporadic case reports, and a single review that summarized 19 case reports published before 2004.8
Method
In this article, we report on a series of 12 patients who developed primary psychiatric symptoms and were subsequently diagnosed with mitochondrial disorders. Diagnosis was initially based on the Thorburn criteria9 and subsequently confirmed with genetic analysis in 11 cases. We describe the psychiatric presentation and clinical phenotypes of our patients, together with their biochemical, neuroradiological, electrophysiological, pathological, and genetic findings. We also comprehensively review all previously-reported cases of patients with mitochondrial disorders presenting with psychiatric symptoms.
Case Series
A mitochondrial disorder was diagnosed in 12 patients with psychiatric symptoms who were evaluated at the Neuropsychiatry and/or Neuromuscular and Neurometabolic Clinic at McMaster University between 1995 and 2010. In all cases, a psychiatric disorder was diagnosed and/or treated by a physician before the diagnosis of a mitochondrial disorder was made. All patients underwent neuroimaging and routine blood work, and the majority had neurophysiological testing, including electroencephalography (EEG) and auditory and visual evoked potentials; 11 patients had muscle biopsies. All patients had molecular genetic analysis, and a pathogenic mutation was identified in 11 of the 12 cases. This study was approved by the St. Joseph’s Healthcare Hamilton and Hamilton Health Sciences research ethics boards.
Review of the Literature
We reviewed the literature of all reported cases of adult patients (age ≥16 years) with mitochondrial disorders who presented with psychiatric symptomatology that was adequately described and reported to be an aspect of their initial clinical presentation. We excluded cases in which cognitive impairment, delirium, or dementia were the only psychiatric manifestations, or in which psychiatric illness developed subsequent to the diagnosis. Where the histories of multiple family members were reported, the proband was entered as the index case, and the clinical presentations of other family members were included in the family history. When two siblings presented at the same time, both were included as cases. We searched the electronic databases MEDLINE (from 1948 to June 13, 2011), EMBASE (from 1980 to June 13, 2011), and PsycINFO (from 1806 to June 13, 2011), using subject headings or keywords for mitochondrial disorders and psychiatric disorders specific to each database. These included “mitochondrial disorder,” “mitochondria,” “mitochondrial cytopathy,” “mitochondrial disease,” which were combined with the Boolean operator OR and then combined using the term AND with “psychiatry” OR “mental disorders” OR “major depression,” “bipolar disorder,” OR “anxiety,” OR “obsessive-compulsive disorder,” OR “schizophrenia,” OR “psychosis.” We also reviewed bibliographies of retrieved articles, relevant review articles, and book chapters and contacted corresponding authors for missing information. There were no language restrictions, and articles in languages other than English were translated.
Results
Cases of Major Depressive Disorder
Case 1:
A 53-year-old woman, with no formal psychiatric history, presented with a 1-year history of progressively worsening depressed mood, fatigue, problems with concentration, and social anxiety. She was diagnosed with major depressive disorder but episodes of acute neurological dysfunction, characterized by dizziness, staggering, left facial numbness, generalized weakness, headache, and expressive aphasia, as well as subjective concern about cognitive decline, prompted further investigation. On review of symptoms, she endorsed muscle soreness and cramping, constipation, urinary frequency, and decreased hearing. She was a nonsmoker and did not consume alcohol. Her medical history was significant for migraine, Type 2 diabetes mellitus, and a left facial palsy during pregnancy. She was on no medication other than an estrogen patch and multivitamins. Family history was significant for a mother with hearing loss, a sister and a brother with premature stroke and ataxia, and another brother with stroke-like episodes, Parkinson’s disease, and cardiomyopathy. Her daughter was noted to have ptosis, and her son has cognitive impairment and psychiatric problems. On examination, she had impaired fine motor movements, mild dysmetria, hearing impairment, poor balance, and a broad ataxic gait. The remainder of her examination was normal. There was little change over time other than ptosis of her left eye, anterior ischemic optic neuropathy in the right eye, and mild parkinsonism. Lactate and ammonia levels were elevated. An initial electroencephalogram (EEG) showed excess slow activity, and a subsequent EEG showed bursts of irregular slow activity over both hemispheres. Auditory testing showed marked hearing impairment bilaterally (threshold: 51 db R, 43 db L). Visual evoked potentials (EVPs) were within normal limits. Cranial magnetic resonance imaging (MRI) revealed generalized atrophy and multiple lesions in the supratentorial white matter. Bilateral basal ganglia calcification was noted on cranial CT. Muscle biopsy showed increased lipid staining, increased subsarcolemmal glycogen, several ragged red fibers, and a predominance of type 2 fibers, consistent with a mitochondrial myopathy. Genetic analysis was positive for MELAS 3243, an A to G transition mutation at position 3243 of the mitochondrial tRNALeu (UUR) gene. After diagnosis, mitochondrial supplements were initiated, including creatine monohydrate, coenzyme Q10, vitamin C, vitamin E, riboflavin, and alpha lipoic acid.10 At 68 years of age, 15 years after presentation, and on no psychiatric medications, she remains independent and has had no cognitive decline or further episodes of major depression. Her cranial MRI is unchanged.
Case 2:
A 60-year-old woman with a 40-year history of dysthymia, generalized anxiety, and mild (untreated) obsessive-compulsive disorder, became depressed after developing a cardiac arrhythmia and being diagnosed with cardiomyopathy. A year later, when she learned that a brother had been diagnosed with mitochondrial disorder, she was referred to the Neurometabolic Clinic for investigation. At this time, she met criteria for major depression and was noted to have short-term memory impairment, word-finding problems, and a tendency to mis-speak. Her medical history was significant for hearing loss, transient ischemic attacks and stroke-like episodes, cataracts, constipation, gastroesophageal reflux, Raynaud’s phenomenon, and colitis. Family history was positive for a mother with postpartum psychotic depression; a brother and sister with depression, strokes, and seizures; and a daughter with depression. Blood work revealed vitamin B12 and D deficiencies and an elevated resting lactate. Her EEG showed abnormalities over both central regions, with no paroxysmal activity. Auditory EVPs from the right ear were poorly formed, and she had bilateral hearing impairment (threshold: 63 db R, 55 db L). Visual EVPs were delayed on the left. MR spectroscopy showed subtle high signal intensity in the left occipital lobe, with a lactate peak in the same area. The first muscle biopsy was normal, but a second revealed many ragged red fibers, multiple internalized nuclei, crystalline inclusions, increased subsarcolemmal staining, and a predominance of type 1 fibers. Genetic testing was positive for MELAS 3271, with a T to C transition mutation at position 3271 of the mitochondrial tRNALeu (UUR) gene.11 For the past 10 years, on a regimen of mitochondrial supplements, she has remained independent and in stable health. From a psychiatric perspective, courses of treatment with nitrazepam, buspirone, trazodone, venlafaxine, and citalopram did not lead to resolution of her depressive symptoms.
Case 3:
This 41-year-old man presented with a 3-year history of major depression. During treatment with three different antidepressant medications (paroxetine, sertraline, and nortriptyline), he developed foot dystonia and hand tremor. He was switched to alprazolam 6 mg. daily, on which he recovered fully, with no depressive relapses over the ensuing 10 years, despite ongoing medical and neurological problems. A consultation regarding his hand tremor and dystonia led to a diagnosis of Parkinson’s disease (PD), which responded well to dopaminergic medication. Six years after this diagnosis, he developed sleep apnea, dilated cardiomyopathy, cataracts, and panic disorder secondary to worsening dystonia. Family history included a sister with chronic depression and cardiomyopathy; a maternal cousin with an intractable seizure disorder; and primary psychotic illness (schizophrenia and schizoaffective disorder) in his mother, three maternal aunts, and two maternal male cousins. Cranial MRI, neurocognitive testing, and visual evoked potentials were normal. Auditory evoked potentials revealed hearing impairment (threshold: 25 db bilaterally). His interest in the “mitochondrial hypothesis of PD” prompted referral to the Neurometabolic Clinic. Investigations revealed normal serum lactate and low vitamin D. A muscle biopsy showed paracrystalline inclusions, consistent with a mitochondrial cytopathy. Genetic analysis revealed a novel A to T transition mutation at 11081, in the ND4 subunit, which was heteroplasmic. Because of the worsening dystonia, he was referred for insertion of a neurostimulator in the subthalamic nucleus, which has been effective in treating his dystonia and parkinsonism. From a psychiatric perspective, he has continued to take alprazolam and mitochondrial supplements and was psychiatrically well at 10-year follow-up.
Case 4:
This 52-year-old man with a history of mood disorder starting at age 48, presented with major depression and catatonia of 1-month duration associated with significant weight loss. He spent his days in bed, refusing to eat or drink and never speaking spontaneously. Symptoms included low mood, fatigue, weakness, extreme anxiety, paranoia, and delusional guilt about past behavior. His history of depression dated to age 48 and included a serious suicide attempt. At that time, neuropsychiatric testing showed a low-average working memory, low-average recall, and poor executive function. He was treated with paroxetine, trifluoperazine, and lorazepam without effect. His medical history was significant for ptosis, which had been surgically corrected, hearing loss, and first-degree heart block. His father and paternal grandfather had ptosis and dysphagia, and had been diagnosed with possible occulopharyngeal dystrophy (not confirmed genetically). His mother, four maternal aunts, and two nieces were hearing-impaired, and one maternal niece had severe muscle weakness. Our patient was admitted to the psychiatric unit. Routine hematological tests were unremarkable. His catatonia responded incompletely to intramuscular lorazepam,12 after which, a course of electroconvulsive therapy (ECT) was initiated. After six ECT treatments, he developed dysarthria, dysphagia, and profound external ophthalmoplegia, which prompted further investigation. Vitamin D and B12 deficiencies were identified, and serum lactate and ammonia were elevated. Evoked potentials and EEG were normal. Cranial MRI showed periventricular, occipital, and parietal white-matter abnormalities, increased signal in the centrum semiovale and pons, and mild diffuse atrophy. Muscle biopsy identified cytochrome oxidase (COX)-negative fibers, mitochondrial accumulations, increased subsarcolemmal staining, and multiple internalized nuclei, and Southern blotting showed a single 3 Kb mtDNA deletion at 73% heteroplasmy. Over the course of the next decade, despite mitochondrial supplements, he developed a profound frontal lobe dementia and died at age 67.
Case 5:
A 43-year-old man presented with a 5-year history of major depression and somatic delusions about parts of his body being misshapen after a musculoskeletal injury. Examination revealed ataxia, bilateral ptosis, external ophthalmoplegia, and dyskinesia of both lower limbs. He had no previous psychiatric history, but his medical history included hypogonadism and a complex regional pain syndrome of his left leg after the aforementioned injury. Family history was unknown. Hematological investigation revealed elevated lactate and ammonia levels. Cranial MRI showed mild atrophy and a small white-matter lesion in the left frontal lobe that was associated with a lactate peak. Visual evoked potentials showed delays from both eyes; auditory evoked potentials were normal (threshold: 15 db bilaterally). A muscle biopsy revealed scattered ragged red and COX-negative fibers, as well as increased lipid staining. Southern blot revealed a single 7.5-Kb heteroplasmic mtDNA deletion. Our patient did not want psychiatric treatment for his depression and anxiety, and he was lost to follow-up. He died in a motor vehicle accident at age 49.
Case 6:
This 45-year-old Ph.D. student presented with a 2-year history of treatment-resistant psychotic depression characterized by low mood, disturbed sleep, anergia, suicidality, and auditory hallucinations. She also expressed concern about a decline in memory and her general intellectual ability. She had no response to treatment with numerous psychotropic agents and had to leave her academic program and apply for social assistance. Past psychiatric history was significant for major depression dating to age 16, when she was hospitalized and treated with ECT. In her 20s, she was diagnosed with borderline personality disorder.13 Her medical history was significant for short stature and “immunologlobulin therapy” for fatigue and weakness as a child. Family history included a father with bipolar disorder, a mother with presenile dementia, and two sisters with depression and panic disorder. Examination revealed that she had inconsistent and bizarre neurological and neurocognitive findings, suggesting conversion disorder. Cranial MRI, however, was grossly abnormal, with extensive periventricular white-matter abnormalities adjacent to the occipital horns of both lateral ventricles. Lumbar puncture and EEG were normal; auditory evoked responses showed sensorineural hearing loss bilaterally; and visual evoked responses showed delays from the left eye. Blood work revealed elevated lactate, and low B12 and vitamin D levels. On neuropsychological testing, she had a full-scale IQ of only 88, with impaired short-term memory, impoverished word fluency, and a poor fund of general knowledge out of keeping with her academic achievements. The first muscle biopsy showed a single ragged red fiber and increased lipid staining, and a repeat muscle biopsy was normal. Genetic analysis on fibroblasts, however, revealed a MELAS 3271 T>C mutation.11 At this point, all psychotropic medications were tapered and discontinued and replaced with mitochondrial supplements. Over the course of the next several years, she showed dramatic improvement in her psychiatric symptoms and was able to return to work full-time and became socially active. She has been stable for 15 years, and repeat neuropsychological testing 15 years after her presentation showed significant improvement, with a full-scale IQ of 100, with improvements in vocabulary, verbal memory, and visual spatial abilities. Her MRI has remained unchanged.
Case 7:
This 46-year-old married man was admitted to Psychiatry for treatment of depression and anxiety. His medications included paroxetine, amitriptyline, clonazepam, oxycodone, and oxycodone/acetaminophen. He had remote a history of alcohol abuse and previous hospital admissions for major depression at ages 30 and 31, and an inability to work since age 38. His psychosocial history was marked by behavioral disturbance and assessment for attention deficit disorder (ADHD) at age 9, sexual abuse, and his father’s suicide when he was 10. In adolescence, he developed migraine headaches and also had difficulty gaining weight. He reported a history of myoclonic jerking since childhood, which worsened over time. He was never able to sire children, and, at age 43, he developed a rather precipitous hearing impairment and congestive heart failure, secondary to cardiomyopathy. He ceased alcohol use, but, over the next 3 years, developed diplopia, ptosis, tremor, constipation alternating with diarrhea, seizures, cataracts, stroke-like episodes, deafness, back pain, muscle cramping, worsening fatigue, and muscle weakness. Neurological examination revealed oral-buccal dyskinesia, jerking movements of his hands and feet, parkinsonism, muscle weakness, and decreased vibration sensation. Lactate levels were elevated, at 4.0 and 4.9, and a muscle biopsy showed ragged red fibers and paracrystalline inclusions. White-matter lesions in the frontal lobes and increased signal in the pons were noted on cranial MRI. Mitochondrial DNA sequencing revealed a novel 760 A>G sequence variant. At age 51, his diplopia, myoclonic jerking, and back pain had all worsened, and he developed progressive external ophthalmoplegia and dysphagia. At that time, his daily medications included paroxetine 40 mg, quetiapine 300 mg, clonazepam 1.5 mg, modafinil 200 mg, propranolol 120 mg, vitamin D 1,000 IU, and mitochondrial supplements. He remained unable to work and chronically depressed.
Case of Anorexia Nervosa
Case 8:
A 21-year-old, previously healthy young woman presented in her final month of pregnancy because of aversion to food, failure to gain weight, and intrauterine fetal growth retardation. She was admitted with a tentative diagnosis of anorexia nervosa; labor was induced, and a female infant was delivered preterm. She developed postpartum depression and, over the next year, had recurrent nausea and vomiting, severe abdominal pain, malnutrition, and paresthesias in all four limbs. She spoke of her use of food intake to manipulate others. Her body mass index (BMI) was 13. Her psychosocial and educational histories were unremarkable. The family psychiatric history was positive for depression in her mother. Medically, she had a brother with bowel problems and inability to gain weight, and an otherwise-healthy sister, also with an inability to gain weight. Gastroenterological investigations revealed ileal diverticulitis, and, 1 year after the birth of her daughter, she developed an obstruction requiring bowel resection. Although there was ongoing concern that she was inducing vomiting, she also clearly had gastrointestinal dysmotility. EMG revealed severe sensory-motor demyelinating neuropathy with axonal loss. Over the next 2 years, she developed recurrent, painful bowel obstructions necessitating treatment with opiates and intermittent parenteral nutrition. Her temperament changed, with emotional lability, periods of confusion, and difficulty with word-finding. She also developed bilateral ptosis and seizures. Cranial imaging showed extensive bilateral white-matter disease, with large confluent lesions and a focal small area of abnormal signal in the right substantia nigra. Lumbar puncture was significant for elevated CSF protein. Blood testing showed high thymidine levels and low thymidine phosphorylase activity, leading to a diagnosis of mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) syndrome. At 5-year follow-up, she showed deterioration, with myoclonus, dysarthria, ptosis, profound peripheral neuropathy with bilateral foot-drop, and episodes of delirium associated with sepsis, electrolyte disturbances, and lactic acidosis, with lactate levels over 4 on occasion.
Cases of Bipolar Affective Disorder With Atypical Features
Case 9:
This 46-year-old mother of two children presented with a 20-year history of treatment-resistant bipolar affective disorder and a 6-month history of progressively worsening ataxia. At the time of presentation, she was taking valproic acid and olanzapine, both of which were withdrawn because of a lack of effect and weight gain with the development of diabetes. Her medical history was significant for fatigue, muscle weakness, Type 2 diabetes mellitus, bilateral hearing loss, lipoma removal, and premature ovarian failure. On neurological examination she had horizontal nystagmus, impaired vibration sense, ataxia, and a positive Romberg test. Her family psychiatric history was significant for a mother with a postpartum psychotic depression and a brother, sister and daughter with depression. The family medical history was positive for lipoma removal in her maternal grandmother, mother, and brother; type 2 diabetes in her sister and a maternal uncle; migraine in her daughter; and hearing loss and type 2 diabetes in her son. Investigation revealed an elevated serum lactate. Auditory evoked potentials showed significant hearing loss. Neuropsychological testing showed problems with memory and executive functioning, and her overall IQ was in the 45th percentile. Cranial MRI revealed five small, discrete, white-matter lesions in the parietal lobes and right frontal lobe as well as a few scattered hyperintensities in deep white matter. Muscle biopsy showed increased lipid staining. Genetic analysis demonstrated a G > A transition mutation at position 8363 of mtDNA. At 3-year follow-up, she continued to have mood lability, and her insight and judgment remain poor. She was treated with mitochondrial supplements, and her psychiatric symptoms most recently are managed with lithium, psychoeducation, exercise, and supportive psychotherapy. She continues to live independently.
Case 10:
This 41-year-old woman presented to our inpatient psychiatric service with psychosis, behavioral disinhibition, poor judgment, and lack of insight. She had first been hospitalized at age 17, at which time she was diagnosed with bipolar affective disorder. She had a remote history of alcohol abuse and, at ages 24 and 34, received full courses of ECT for depression. She is of short stature, and, over time, developed seizures, migraine, hypothyroidism, a left bundle-branch heart block, and a left frontal lobe cerebral infarction. On neurological examination, she had oral buccal dyskinesia, proximal muscle weakness, hyperreflexia, extensor plantar responses bilaterally, a positive Romberg sign, impaired tandem gait, and decreased vibration sense. Her family history was significant for a father who died of premature stroke; a mother, sister, brother, and maternal niece with seizure disorders; and three siblings with cardiac problems. Hematological investigation showed elevated lactate and low levels of vitamin D. Repeat EEGs have shown diffuse excess slow activity. Visual evoked potentials were normal, but auditory evoked potentials showed impaired hearing (threshold: 25db R, 16db L). Neuropsychiatric testing at age 41 demonstrated a Verbal IQ of 80 and Performance IQ of less than 70. Cranial MRI showed multiple periventricular and subcortical high signal intensities in the frontal and parietal lobes regions. MR spectroscopy showed a lactate peak. A muscle biopsy had prominent lipid staining and mild nonspecific changes, including focal internalized nuclei and partial mild type 2 fiber atrophy. Mitochondrial genome sequencing revealed several polymorphisms, but a definitive mutation could not be identified. Over 17-year follow-up, she has remained persistently psychotic, with perceptual abnormalities, mood lability, and aggression, as well as cognitive decline in keeping with frontal lobe dementia. At age 52, she was admitted to a long-term care facility because of her family’s inability to care for her.
Cases of Treatment-Resistant Obsessive-Compulsive Disorder (OCD)
Case 11:
This 24-year-old single man presented with a 5-year history of obsessions and compulsions. He had a chronically low mood and had become more depressed after the death of his grandparents 2 years before presentation. His psychiatric history was significant for obsessions, compulsions, and generalized anxiety beginning in his early teens. His medical history was unremarkable except for lipomas and a remote umbilical hernia repair. On presentation, he had visible muscle wasting and multiple lipomas on his back and neck bilaterally. The presence of lipomas, as well as a family history of lipomas in his grandmother and mother, who was diagnosed with MERRF 8344, prompted consideration of a mitochondrial disorder. His serum lactate level was elevated, and his visual evoked potentials showed delays bilaterally. A cranial CT was normal. Mitochondrial DNA mutation analysis was positive for an A > G transition mutation at position 8344 of mtDNA. His OCD was treated with citalopram, fluoxetine, and behavioral treatment without significant improvement. Over the next 5 years, he developed muscle wasting, dysarthria, dysphagia, dystonia, and optic atrophy, and he died at age 29 from respiratory failure.
Case 12:
A 48-year-old married woman, looking significantly older than her stated age, and with bilateral hearing aids, presented with an 8-month history of obsessive-compulsive behavior related to fears of contamination and fire. She engaged in excessive checking, hand-washing, and cleansing rituals. She had her first admission for depression at age 42 and had previously been diagnosed with anorexia nervosa because of her very low weight, although she denied restricting her intake or having a disturbed body image. Her OCD symptoms had been treated with several different SSRI antidepressant agents, with little response. Psychiatric history also included a “nervous breakdown” at age 42 and attempted suicide at age 44. Her medical history was significant for bilateral hearing loss, short stature, recurrent miscarriages, and severe constipation with pseudo-obstruction, Type 2 diabetes mellitus, and optic atrophy. She was a smoker with no history of alcohol or drug abuse. She had one daughter with a learning disability, attention-deficit hyperactivity disorder, and anxiety disorder. Her mother had short stature and a premature stroke, and a brother had hearing loss. Neuropsychological testing showed impairments in receptive language, dyscalculia, mild constructional dyspraxia, and poor executive functioning. Numerous frontal lobe signs were elicited, including a strongly positive glabellar sign, bilateral palmomental reflexes, snout reflex, and paratonia. Abnormal neurological findings included left-sided ptosis, limited extraocular movements, nystagmus, postural tremor, impaired alternate motion rate, truncal ataxia, proximal muscle weakness, and impaired vibration sense. Lactate was elevated. An EEG was normal; auditory evoked potentials showed bilateral hearing loss, and visual evoked potentials were delayed from the left eye. Cranial MRI showed large symmetric areas of high signal intensity in the pons and several white-matter hyperintensities in the frontal lobes and periventricular regions. Muscle biopsy revealed ragged red fibers, occasional COX-negative fibers, paracrystalline inclusions, and reduction in complex 1 and 3 activity. Genetic analysis was positive for MELAS A3243G. She was started on mitochondrial supplements and withdrawn from all psychotropic medications except fluvoxamine 25 mg. At 10-year follow-up, she is independent with self-care, free of psychiatric symptoms, and stable from a neurological perspective, albeit with frontal lobe features. Her weight remains low.
Review of the Literature
A total of 47 cases of patients with mitochondrial disorders presenting with prominent psychiatric symptoms were identified.14–56 Their psychiatric presentations and diagnoses are summarized in Table 1. Additional information, including detailed clinical descriptions, medical histories, relevant family histories and investigations can be found in the online data supplement.
| Case | Sex | Onset of Psychiatric Symptoms, years | Age at Diagnosis, years | Major Depressive Disorder | Bipolar Affective Disorder | Psychosis | OCD or Anxiety | Cognitive Deterioration | Frontal Lobe Syndrome | Personality Change | Diagnosis | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F | 16 | NR | + | — | + | — | — | — | — | Mitochondrial cytopathy | Ahn et al., 200514 |
| 2 | M | 29 | 36 | — | — | + | — | + | — | — | C3256T | Amemiya et al., 200015 |
| 3 | F | 58 | 58 | — | — | + | — | + | — | — | MELAS 3243 | Apostolova et al., 200516 |
| 4 | M | 25 | 37 | — | — | + | — | — | — | — | MELAS 3243 | Ban et al., 199217 |
| 5 | F | NR | 28 | + | — | + | — | — | — | — | MELAS | Bhuvaneswar et al., 200856 |
| 6 | F | 44 | 44 | — | — | + | — | + | — | — | MELAS 3243 | Clark et al., 199618 |
| 7 | F | NR | 33 | + | — | + | — | — | — | — | C3303T | Campos et al., 200119 |
| 8 | M | 24 | 46 | — | — | + | — | — | — | — | KSS | Desnuelle et al., 198920 |
| 9 | M | 22 | 61 | + | — | + | — | — | — | — | Mitochondrial cytopathy | Gardner et al., 200321 |
| 10 | M | 22 | 61 | + | — | + | — | — | — | — | Mitochondrial cytopathy | Grover et al., 200646 |
| 11 | F | 16 | NR | + | — | + | — | — | — | — | POLG1 mutation | Hopkins et al., 201047 |
| 12 | F | 17 | NR | + | — | + | — | — | — | — | POLG1 mutation | Hopkins et al., 201047 |
| 13 | M | 31 | 35 | — | — | + | — | + | + | — | MELAS 3243 | Inagaki et al., 199722 |
| 14 | M | 27 | 33 | + | — | + | — | + | — | — | MELAS 3274 | Jaksch et al., 200123 |
| 15 | F | 53 | 53 | — | — | + | — | — | — | — | MELAS 3243 | Kaido et al., 199624 |
| 16 | M | 32 | 32 | — | — | + | — | + | — | — | MELAS | Kiejna et al., 200225 |
| 17 | F | 12 | 16 | + | — | + | — | + | — | — | PDHA1 mutation | Koene et al., 200948 |
| 18 | F | 14 | 16 | + | — | — | — | — | — | — | MTND1 mutation | Koene et al., 200948 |
| 19 | F | 16 | 17 | + | — | + | — | — | — | — | POLG1 mutation | Koene et al., 200948 |
| 20 | F | 15 | 16 | + | — | — | — | + | — | — | MTTK mutation | Koene et al., 200948 |
| 21 | M | 34 | 37 | — | — | — | — | — | + | + | MELAS 3243 | Koller et al., 200326 |
| 22 | F | NR | NR | + | — | + | — | + | — | — | POLG mutation | Komulainen et al., 201049 |
| 23 | M | 30 | 30 | — | — | — | + | — | — | — | MELAS 3243 | Lacey et al., 200827 |
| 24 | M | 49 | 51 | — | — | — | + | — | — | — | MELAS 3243 | Lacey et al., 200827 |
| 25 | F | 12 | 63 | + | — | — | — | + | — | — | Mt DNA deletions | Mancuso et al,. 200850 |
| 26 | M | 47 | 52 | + | — | — | — | — | — | MELAS 3243 | Miyaoka et al., 199728 | |
| 27 | F | 37 | 62 | — | — | — | + | — | — | — | MELAS 3243 | Miyaoka et al., 199728 |
| 28 | F | 25 | 69 | — | — | — | + | — | — | — | MELAS 3243 | Miyaoka et al., 199728 |
| 29 | M | 35 | 61 | — | — | — | + | — | — | — | MELAS 3243 | Miyaoka et al., 199728 |
| 30 | F | 23 | 52 | — | — | + | — | + | — | — | MELAS | Odawara et al., 199731 |
| 31 | M | 22 | 23 | + | — | — | + | — | — | — | MELAS 3243 | Onishi et al., 199732 |
| 32 | F | 27 | 47 | — | — | + | — | — | — | — | MELAS 3243 | Prayson et al., 199833 |
| 33 | M | 30 | 47 | + | — | — | — | + | — | — | POLG mutation | Rantamaki et al., 200152 |
| 34 | M | 28 | 34 | — | — | + | — | — | + | + | MELAS 3243 | Sartor et al., 200235 |
| 35 | F | 48 | 48 | — | — | + | — | + | — | — | MELAS 3243 | Shinkai et al., 200037 |
| 36 | F | NR | 44 | — | + | + | — | — | — | — | ANT1 mutation | Siciliano et al., 200353 |
| 37 | M | 21 | 36 | — | — | + | — | — | — | MELAS 3243 | Spellberg et al., 200138 | |
| 38 | F | 23 | 44 | — | + | — | — | + | — | — | KSS | Stewart and Naylor, 199039 |
| 39 | F | 19 | 29 | + | — | + | — | + | — | AD PEO, Twinkle mutation | Suomalainen et al., 199240 | |
| 40 | M | 25 | 35 | — | — | + | — | + | — | — | MELAS | Suzuki et al., 198941 |
| 41 | F | 29 | NR | + | — | + | — | — | — | — | MELAS 3251 | Sweeney et al., 199343 |
| 42 | F | 25 | 39 | — | — | — | — | — | — | — | MELAS 3251 | Sweeney et al., 199343 |
| 43 | F | 22 | 27 | — | — | + | — | + | — | — | MELAS 3243 | Thomeer et al., 199844 |
| 44 | M | 23 | 39 | NR | NR | NR | NR | NR | NR | NR | POLG mutation | Van Goethem et al., 200451 |
| 45 | F | 37 | 52 | + | — | + | — | — | — | — | POLG mutation | Verhoeven et al., 201154 |
| 46 | M | 29 | 37 | — | — | + | — | + | — | — | MELAS | Yamazaki et al., 199145 |
| 47 | F | 57 | NR | + | — | + | — | + | — | — | MTTF mutation | Young et al., 201055 |
| Mean/Total | 29 | 41 | 20 | 2 | 30 | 6 | 18 | 4 | 2 | |||
Discussion
There is increasing awareness that psychiatric symptomatology is common among patients with known mitochondrial cytopathies. Recent reports indicate that 70% of adult mitochondrial-disorder patients will have evidence of major mental illness at some point in their lives,57 and depressive behavior has been reported in 50% of children with a mitochondrial disorder.58 To the best of our knowledge, this is the first comprehensive description of patients with mitochondrial disorders presenting with psychiatric illness. Based upon our detailed case series and review of the cases described in the literature, we summarize the psychiatric presentation of mitochondrial disorders and discuss the relevant diagnostic considerations and course of illness.
Psychiatric Features
The psychiatric features of the 12 patients in our series are presented in Table 2. The age at onset of psychiatric symptoms was taken to be the first time a psychiatric disorder was diagnosed and/or treated by a physician, and this ranged from 15 to 53 years. The age at diagnosis of a mitochondrial disorder ranged from 24 to 54 years. In all cases, the onset of psychiatric symptoms predated the diagnosis of a mitochondrial disorder by 13 years, on average. Of the 12 patients, 11 met diagnostic criteria for treatment-resistant psychiatric illness, as defined by failure to achieve remission with two adequate trials of psychotropic medications,59 and three patients (Patients 4, 6, and 9) showed deterioration while taking psychotropic medications, with improvement when they were discontinued. The major presentation for seven patients (Patients 1–7) was major depressive disorder; three of the seven (Patients 4, 5, and 6) also had psychotic features. Bizarre episodes of neurological dysfunction in Patients 1 and 6 raised the possibility of malingering or a conversion disorder. One patient (Patient 8) was diagnosed with anorexia nervosa; two patients (Patients 9 and 10) presented with bipolar disorder; and two others (Patients 11 and 12) met diagnostic criteria for OCD; 11 patients had comorbid psychiatric conditions, including anxiety disorders, substance abuse, borderline personality disorder, and catatonia.
| Patient/Sex | 1/F | 2/F | 3/M | 4/M | 5/M | 6/F | 7/M | 8/F | 9/F | 10/F | 11/M | 12/F | Mean Total |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age at onset psychiatric disorder, years | 53 | 47 | 41 | 48 | 38 | 16 | 30 | 20 | 20 | 17 | 15 | 42 | 32 |
| Age at diagnosis of mitochondrial disorder, years | 54 | 50 | 54 | 52 | 43 | 45 | 46 | 24 | 46 | 54 | 24 | 48 | 45 |
| Family psychiatric history | + | — | + | — | — | + | + | — | + | — | — | + | 6 |
| Treatment-resistant illness | + | + | + | + | + | + | — | + | + | + | + | + | 11 |
| Deterioration on psychotropic medications | — | — | — | + | — | + | — | — | + | — | — | — | 3 |
| Mood disorder | 12 | ||||||||||||
| Major depressive disorder | + | + | + | + | + | + | + | + | — | — | + | + | 10 |
| With psychotic features | — | — | — | + | + | + | — | — | — | — | — | — | 3 |
| Bipolar Affective disorder | — | — | — | — | — | — | — | — | + | + | — | — | 2 |
| With psychotic features | — | — | — | — | — | — | — | — | — | + | — | — | 1 |
| Anxiety disorder | 7 | ||||||||||||
| Generalized anxiety disorder | — | + | + | — | — | — | + | — | — | — | + | + | 5 |
| Social anxiety | + | — | — | — | — | — | — | — | — | — | + | + | 3 |
| Obsessive-compulsive disorder | — | + | — | — | — | — | — | — | — | — | + | + | 3 |
| Panic disorder | — | — | + | — | — | — | — | — | + | — | — | — | 2 |
| Cognitive disorder | 6 | ||||||||||||
| Cognitive deterioration | + | + | — | + | — | + | — | — | — | + | — | + | 6 |
| Frontal lobe syndrome | — | — | — | + | — | — | — | — | — | + | — | + | 3 |
| Other | |||||||||||||
| Anorexia nervosa | — | — | — | — | — | — | — | + | — | — | — | + | 2 |
| Borderline personality disorder | — | — | — | — | — | + | — | — | — | — | — | — | 1 |
| Catatonia | — | — | — | + | — | — | — | — | — | — | — | — | 1 |
| Substance abuse or dependence | — | — | — | — | — | — | + | — | — | — | — | — | 1 |
In the 47 cases reported in the literature, the average age at onset of psychiatric symptoms predated the average age at diagnosis of a mitochondrial disorder by over a decade (Table 1). The most common presentation was depression (N=18) with psychotic features evident in 12. The second most common presenting symptom was psychosis (N=17), diagnosed as schizophrenia, schizoaffective disorder, and delusional disorder. Other diagnoses included cognitive deterioration (N=16), anxiety disorders (N=9), bipolar disorder (N=3), and frontal lobe syndrome (N=3). In seven cases (15%) there was specific mention of treatment-resistance, and four patients (9%) were noted to have marked deterioration after psychotropic medications were introduced (see online Data Supplement).
Physical Features and Past Medical History
The majority of patients in our series (N=12) either presented with a history of, or developed, medical problems affecting multiple organ systems (Table 3). The most common conditions were muscle weakness or atrophy (N=10), hearing loss (N=8), and fatigue (N=8). Other common medical conditions included dysphagia, constipation, Type 2 diabetes mellitus, migraine, and stroke/stroke-like episodes. Information on the medical history of the cases in the literature can be found in the Data Supplement. The most common nonpsychiatric features in the 47 cases in the literature were muscle weakness or atrophy (N=23), seizure disorder (N=15), migraine or headache (N=15), hearing loss (N=14), and short stature (N=12). Other features included Type 2 diabetes mellitus (N=11), severe constipation often with ileus (N=7), ataxia (N=6), dysarthria (N=6), strokes or stroke-like episodes (N=6), Wolf-Parkinson-White syndrome (N=5), ophthalmoplegia (N=5), ptosis (N=5), cardiomyopathy (N=4), cardiac conduction defect (N=3), and abnormal movements (N=4). Most had multi-organ system involvement.
| Patient/Sex | 1/F | 2/F | 3/M | 4/M | 5/M | 6/F | 7/M | 8/F | 9/F | 10/F | 11/M | 12/F | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mitochondrial disorder diagnosis | MELAS 3243 | MELAS 3271 | Novel mutation: 11081A>T | 3.3kb deletion | CPEO 7.5kb deletion | MELAS 3271 | Novel mutation: 760A>G | MNGIE | MERRF 8363G>A | Unknown | MERRF 8344 | MELAS 3243 | Total |
| Medical Histories | |||||||||||||
| Muscle weakness or atrophy | + | — | — | + | + | + | + | + | + | + | + | + | 10 |
| Hearing loss | + | + | — | + | — | + | + | + | + | — | — | + | 8 |
| Fatigue | + | + | — | + | + | + | + | + | + | — | — | — | 8 |
| Dysphagia | — | — | — | + | + | — | + | + | + | — | + | — | 6 |
| Constipation | + | + | — | — | — | — | + | + | — | — | — | + | 5 |
| Stroke/stroke-like episodes | + | + | — | — | — | + | + | — | — | + | — | — | 5 |
| Type 2 diabetes | + | — | — | — | — | + | — | — | + | — | — | + | 4 |
| Migraines or headaches | + | — | — | — | — | — | + | + | — | + | — | — | 4 |
| Cardiac conduction abnormality | — | — | + | + | — | — | — | — | — | + | — | — | 3 |
| Short stature | — | — | — | — | — | + | — | — | — | + | — | + | 3 |
| Dysarthria | — | — | — | + | — | — | — | — | — | + | + | — | 3 |
| Cardiomyopathy | — | + | + | — | — | — | + | — | — | — | — | — | 3 |
| Seizure disorder | — | — | — | — | — | — | + | + | — | + | — | — | 3 |
| Lipomas | — | — | — | — | — | — | — | — | + | — | + | — | 2 |
| Dystonia | — | — | + | — | — | — | — | — | — | — | + | — | 2 |
| Arrhythmia | — | + | — | — | — | — | — | — | — | — | — | — | 1 |
| Recurrent miscarriages | — | — | — | — | — | — | — | — | — | — | — | + | 1 |
| Sleep apnea | — | — | + | — | — | — | — | — | — | — | — | — | 1 |
| Premature ovarian failure | — | — | — | — | — | — | — | — | + | — | — | — | 1 |
| Physical Examination | |||||||||||||
| Ataxia | + | — | — | + | + | + | — | — | + | + | + | — | 7 |
| Impaired vibration sense | + | — | — | + | — | — | + | — | + | + | + | + | 7 |
| Ptosis | + | — | — | + | + | — | + | + | — | — | — | + | 6 |
| Ophthalmoplegia | — | — | — | + | + | — | + | + | — | — | — | + | 5 |
| Babinski sign | — | — | — | + | — | + | — | — | — | + | + | — | 4 |
| Positive Romberg sign | — | — | — | + | — | — | — | — | + | + | — | — | 3 |
| Retinitis pigmentosa | + | — | — | + | — | — | — | — | — | — | — | — | 2 |
| Optic atrophy | + | — | — | — | — | — | — | — | — | — | + | — | 2 |
| Parkinsonism | + | — | + | — | — | — | — | — | — | — | — | — | 2 |
| Cataracts | — | + | + | — | — | — | + | — | — | — | — | — | 3 |
| Myoclonus or dyskinesia | — | — | — | — | + | — | + | — | — | + | — | — | 3 |
| Nystagmus | — | — | — | — | — | — | — | — | + | — | — | + | 2 |
| Hyperreflexia | — | — | — | — | — | + | — | — | — | + | — | — | 2 |
Laboratory Investigations
In our series, 10 patients had elevated lactate, a marker of impaired oxidative metabolism, although 4 of these initially had a normal level. Of the 47 cases in the literature, lactate was measured in 29 and found to be elevated in 21 (72%). Given the potential for vitamin deficiency to induce neuropsychiatric symptoms,60,61 it is noteworthy that, in our series, 3/7 had low serum levels of vitamin B12 (<133 pmol/L) and 5/5 had low vitamin D (<80 nmol/L, with 4 patients <50 nmol/L; values were not available for the other cases).
Neuroradiologic Findings
Cranial MRI was abnormal in 10 of the 11 patients that we scanned (Table 4). The most common findings were white-matter abnormalities (N=10) and atrophy out of keeping with age (N=4). Multiple foci of increased signal and hyperintensities were observed in the periventricular regions (N=4), frontal lobes (N=5), parieto-occipital lobes (N=4), and pons (N=3). Three patients4,7,9 had confluent white-matter abnormalities. One of the two patients who had a CT scan had basal ganglia calcifications. Four showed lactate peaks on spectroscopy, generally associated with an area of high signal intensity. In the 28 cases in the literature that reported performing an MRI, the most common findings were white-matter lesions (N=10), cerebral or cerebellar atrophy (N=9), evidence of ischemia or an old infarct (N=3), and basal ganglia calcifications or hyperintensities (N=6). Five of the 28 cases had a normal MRI. Findings in the 7 cases that reported CT results included hypodensities or evidence of infarction (N=2), basal ganglia hyperintensities or infarction (N=2), and cerebellar atrophy (N=1); 3 had a normal cranial CT scan. SPECT scans were reported for 9 patients, with 6 showing decreased cerebral blood flow, mainly in the parieto-occipital areas.
| Patient/Sex | 1/F | 2/F | 3/M | 4/M | 5/M | 6/F | 7/M | 8/F | 9/F | 10/F | 11/M | 12/F | Total/Mean |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Laboratory Investigations | |||||||||||||
| Elevated lactate | + | + | — | + | + | + | + | — | + | + | + | + | 10/12 |
| Maximum lactate (N: 0.7–2.2 mmol/L) | 4.4 | 6.8 | — | 7.9 | 6.7 | 4.7 | 4.9 | — | 2.5 | 4.4 | 6.1 | 2.7 | 5.1 |
| Elevated ammonia | + | — | — | + | + | — | — | — | — | — | — | — | 3/12 |
| Low pyruvate | — | — | — | — | — | — | — | — | — | + | — | + | 2/12 |
| Neuroimaging | |||||||||||||
| MRI: | |||||||||||||
| Normal | — | — | + | — | — | — | — | — | — | — | X | — | 1/11 |
| Atrophy | + | — | — | + | + | — | — | — | — | + | X | — | 4/11 |
| White-matter lesions | + | + | — | + | + | + | + | + | + | + | X | + | 10/11 |
| Lactate on MRS | — | + | — | — | + | + | — | — | — | + | X | — | 4/11 |
| CT: | |||||||||||||
| Normal | — | X | X | X | X | X | X | X | X | X | + | X | 1/2 |
| Atrophy | — | X | X | X | X | X | X | X | X | X | — | X | 0/2 |
| Basal ganglia calcifications | + | X | X | X | X | X | X | X | X | X | — | X | 1/2 |
| Electrodiagnostics | |||||||||||||
| EEG | A | A | X | X | X | N | X | A | X | A | X | N | 4A/6 |
| Auditory EVP | A | A | X | X | N | A | X | X | A | A | X | A | 6A/7 |
| Visual EVP | A | A | X | X | A | A | X | X | N | N | A | A | 6A/8 |
| Neuropsychiatric testing | X | X | N | A | X | A | X | X | A | A | X | A | 5A/6 |
| Muscle biopsy | |||||||||||||
| Ragged red fibers | + | + | — | + | + | + | + | X | — | — | X | + | 7/10 |
| Cox-negative fibers | — | — | — | + | + | — | — | X | + | — | X | + | 4/10 |
| Respiratory chain complex function | N | N | N | X | X | N | N | X | A | N | X | A | 2A/8 |
Electrophysiological Investigations
The majority of patients in our series had an EEG, visual EVPs, and auditory EVPs (Table 4). In two patients with seizure disorder (Patients 8 and 10), the EEG was abnormal, with both showing fluctuating slow activity and focal sharp waves. Auditory EVPs were abnormal in five patients with hearing loss, not available for the remaining two patients with hearing loss, and normal in only one. Visual EVPs were abnormal in six of eight patients, often with significant latencies in the P100 and N120 components unilaterally or bilaterally. Electrophysiological investigations were not reported in the majority of the cases in the literature.
Muscle Biopsies
Muscle biopsies were performed on 10 of our 12 patients and were abnormal in every case (Table 4). For two patients (#2 and #6), this represented the second biopsy, after an initially normal result. Seven patients (58%) had “ragged red fibers,” a pathognomonic finding in mitochondrial disorders, caused by the accumulation of abnormal mitochondria under the sarcolemmal membrane. Four patients (40%) had COX-negative fibers. Other abnormalities seen on muscle biopsy included increased lipid staining, paracrystalline inclusions within mitochondria, internalized nuclei, and type 2 fiber atrophy. In the 47 cases described in the literature, muscle biopsies were reported in 26, with ragged red fibers seen in 21 (81%) and COX-negative fibers in 8 (31%). Overall, our findings on muscle biopsy are consistent with those reported in the literature.
Correlation Between Clinical Phenotype and Genotype
In our series, the most common diagnosis was MELAS, two with the 3243 mutation and two with the 3271 mutation. Other diagnoses included MERRF 8363, MERRF 8344, CPEO with a 7.5 kb deletion, a 3.3 kb deletion, MNGIE, and two novel mutations. No clear genotype/psychiatric phenotype relationship emerged. In the literature, as well, MELAS mutations have, to-date, been the most likely gene abnormality found in patients presenting with psychiatric symptoms. This may change as more mitochondrial and nuclear mutations are identified and genetic screening allows for greater detection of rare and novel mutations.
Diagnostic and Treatment Considerations
A schema for diagnosing mitochondrial disorders in patients with psychiatric illness is presented in Figure 1. It is important to include an underlying mitochondrial disorder in the differential diagnosis of patients with psychiatric illness in the context of a particular constellation of findings on presentation and history. The majority of cases in our series, and in the literature, had personal and family histories of multiple medical symptoms, including muscle weakness, hearing loss, fatigue, dysphagia, constipation, Type 2 diabetes mellitus, migraine, and stroke-like episodes. From a psychiatric perspective, several patients had atypical aspects to their presentation, and did not conform to strict DSM-IV diagnostic categories. A maternal pattern of inheritance was present for most patients. Deterioration on psychotropic medications, which occurred in several of our patients, may be an important clue to the diagnosis. Several psychotropic medications impair mitochondrial functioning and therefore may worsen symptoms.62 Typical and atypical antipsychotics impair complex I of the mitochondrial respiratory chain; selective serotonin reuptake inhibitors (SSRIs) and tricyclic antidepressants inhibit the mitochondrial respiratory chain and oxidative phosphorylation; and valproic acid can cause secondary impairment of mitochondrial functioning through the induction of carnitine deficiency.63–66 Furthermore, many drugs also have side effects that may contribute to or exacerbate medical conditions associated with mitochondrial disorders.62 For example, many psychotropic medications lower the seizure threshold; tricyclic medications may contribute to cardiac conduction abnormalities; SSRIs may worsen headaches; medications with anticholinergic properties may compound constipation and cognitive decline; and medications such as atypical antipsychotics that are associated with a metabolic syndrome may increase the risk of diabetes mellitus. Therefore, making the diagnosis has particularly important treatment implications for patients with psychiatric illness.
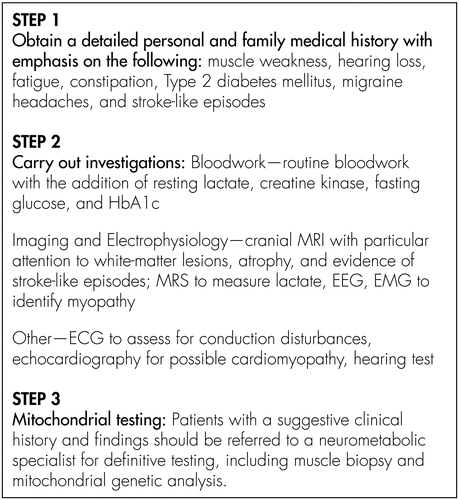
Several findings can be supportive of the diagnosis of a mitochondrial disorder. Extensive supratentorial white-matter lesions and atrophy out of keeping with age were the most common findings on neuroimaging. Lactate level, measured using magnetic resonance spectroscopy (MRS), was also helpful in making the diagnosis in several cases. All patients in our series, and the majority of cases in the literature, had abnormal muscle biopsies suggestive of a mitochondrial cytopathy. Multiple biopsies may be necessary in some cases because of heteroplasmy, whereby one area of the muscle has a high load of mitochondrial mutations, but another has only healthy mitochondria.6,67 An elevated serum lactate level may suggest the disorder, although this, again, may only be evident on repeat testing.
In our series, seven Patients 1, 3, 5, 6, 9, 10, 12 initially presented to the neuropsychiatry service and five Patients 2, 4, 7, 8, 11 initially presented to the neurometabolic and neuromuscular clinic at McMaster University. Given that patients may present to family physicians, psychiatrists, neurologists, and neurometabolic or neuromuscular specialists, it is important that all physicians be aware of the psychiatric presentation of mitochondrial disorders. In our experience, it is often the pattern of medical, neurological, and psychiatric features in both personal and family history that suggests a mitochondrial disorder, which reinforces the importance of taking a thorough history. Once the diagnosis is considered, referral to a neurometabolic or neuromuscular specialist is useful because genetic testing requires sophisticated knowledge of mitochondrial genetics, and often multiple tests are required before a definitive diagnosis is reached.68
Course of Illness
The patients in our series were followed over a median time of 10 years. From a psychiatric, cognitive, and neurological perspective, the majority (58%) remained stable on minimal or no psychotropic medications. This may be attributable in part to our use of mitochondrial supplements, including coenzyme Q10, creatine monohydrate, alpha lipoic acid, vitamin E, vitamin C, and riboflavin. Another factor may be the reduction or discontinuation of psychotropic drugs, some of which can exacerbate the underlying mitochondrial dysfunction. Although the course of illness was not captured in many cases in the literature, eight studies reported an improvement of psychiatric symptoms with coenzyme Q10 supplementation (dose range: 60 mg to 150 mg/day),18,20,21,25,29,32,37,45 and three with the antioxidant idebenone.22,32,45
Limitations
Because our series was retrospective, we must rely on information gathered at the time of the assessments. Fortunately, our patients routinely underwent extensive examination and investigation and were followed closely, so that relevant clinical and diagnostic information has been available for the majority of cases. Patients seen by our service were not consecutively evaluated for a mitochondrial disorder, and therefore the diagnosis may have been missed in some cases; as a result of which, the sample may not be representative. Also, reliable conclusions about treatment cannot be drawn from case series because patients were not randomized or blinded. Our review of the cases reported in the literature was performed using a comprehensive search strategy and is the most complete review of the psychiatric presentation of mitochondrial disorders to-date. However, case reports are subject to reporting bias and therefore may not be entirely representative. Despite these limitations, case reports and case series remain one of the most useful methods for describing clinical phenomena and increasing awareness about the constellation of clinical features that should raise suspicion of a particular disorder. Also, the information in this case series and review can be used to shape future studies on the psychiatric manifestations of mitochondrial disorders.
To our knowledge, this is the first case series of unrelated patients with mitochondrial disorders presenting with psychiatric symptoms. Our series, in combination with cases described in the literature, provides valuable information about the clinical patterns that should prompt consideration of these disorders in patients presenting with psychiatric illness. The diagnosis of a mitochondrial disorder has important treatment implications for patients with psychiatric illness because many psychotropic medications inhibit mitochondrial functioning, and mitochondrial supplements may improve symptoms. Therefore, it is important that physicians be familiar with the psychiatric presentation of mitochondrial disorders and maintain a high index of suspicion for the diagnosis.
1 : Molecular genetic aspects of human mitochondrial disorders. Annu Rev Genet 1995; 29:151–178Crossref, Medline, Google Scholar
2 : Mitochondrial Disorders Overview, in GeneReviews. Edited by Pagon RABird TDDolan CR. Seattle, WA, Univ. of Washington, 1993Google Scholar
3 : Mitochondrial DNA mutations in human disease. Am J Med Genet 2001; 106:18–26Crossref, Medline, Google Scholar
4 : Prevalence of mitochondrial DNA disease in adults. Ann Neurol 2008; 63:35–39Crossref, Medline, Google Scholar
5 : Mitochondrial respiratory-chain diseases. N Engl J Med 2003; 348:2656–2668Crossref, Medline, Google Scholar
6 : Metabolic myopathies: update 2009. J Clin Neuromuscul Dis 2009; 10:97–121Crossref, Medline, Google Scholar
7 : Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature 1988; 331:717–719Crossref, Medline, Google Scholar
8 : Review of the literature on major mental disorders in adult patients with mitochondrial diseases. Psychosomatics 2006; 47:1–7Crossref, Medline, Google Scholar
9 : Diagnostic criteria for respiratory chain disorders in adults and children. Neurology 2002; 59:1406–1411Crossref, Medline, Google Scholar
10 : Beneficial effects of creatine, CoQ10, and lipoic acid in mitochondrial disorders. Muscle Nerve 2007; 35:235–242Crossref, Medline, Google Scholar
11 : Clinical, physiological, and histological features in a kindred with the T3271C MELAS mutation. Muscle Nerve 1998; 21:25–33Crossref, Medline, Google Scholar
12 : Catatonia and its treatment. Schizophr Bull 2010; 36:239–242Crossref, Medline, Google Scholar
13 : The psychiatric manifestations of mitochondrial disorders: a case and review of the literature. J Clin Psychiatry 2012; 73:506–512Crossref, Medline, Google Scholar
14 : Risperidone-induced psychosis and depression in a child with a mitochondrial disorder. J Child Adolesc Psychopharmacol 2005; 15:520–525Crossref, Medline, Google Scholar
15 : Psychosis and progressing dementia: presenting features of a mitochondriopathy. Neurology 2000; 55:600–601Crossref, Medline, Google Scholar
16 : Deep white matter pathologic features in watershed regions: a novel pattern of central nervous system involvement in MELAS. Arch Neurol 2005; 62:1154–1156Crossref, Medline, Google Scholar
17 : An autopsy case of mitochondrial encephalomyopathy (MELAS) with special reference to extra-neuromuscular abnormalities. Acta Pathol Jpn 1992; 42:818–825Medline, Google Scholar
18 : MELAS: Clinical and pathologic correlations with MRI, xenon/CT, and MR spectroscopy. Neurology 1996; 46:223–227Crossref, Medline, Google Scholar
19 : Mitochondrial myopathy, cardiomyopathy and psychiatric illness in a Spanish family harbouring the mtDNA 3303C > T mutation. J Inherit Metab Dis 2001; 24:685–687Crossref, Medline, Google Scholar
20 : [Kearns-Sayre syndrome: mitochondrial encephalomyopathy caused by deficiency of the respiratory chain]. Rev Neurol (Paris) 1989; 145:842–850Medline, Google Scholar
21 : Alterations of rCBF and mitochondrial dysfunction in major depressive disorder: a case report. Acta Psychiatr Scand 2003; 107:233–239Crossref, Medline, Google Scholar
22 : Psychiatric symptoms in a patient with diabetes mellitus associated with point mutation in mitochondrial DNA. Biol Psychiatry 1997; 42:1067–1069Crossref, Medline, Google Scholar
23 : A mutation in mt tRNALeu(UUR) causing a neuropsychiatric syndrome with depression and cataract. Neurology 2001; 57:1930–1931Crossref, Medline, Google Scholar
24 : Alzheimer-type pathology in a patient with mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS). Acta Neuropathol 1996; 92:312–318Crossref, Medline, Google Scholar
25 : Psychiatric symptoms in a patient with the clinical features of MELAS. Med Sci Monit 2002; 8:CS66–CS72Medline, Google Scholar
26 : Persistent organic personality change as rare psychiatric manifestation of MELAS syndrome. J Neurol 2003; 250:1501–1502Crossref, Medline, Google Scholar
27 : Psychiatric comorbidity and impact on health service utilization in a community sample of patients with epilepsy. Epilepsia 2009; 50:1991–1994Crossref, Medline, Google Scholar
28 : Mental disorders in diabetic patients with mitochondrial transfer RNA(Leu) (UUR) mutation at position 3243. Biol Psychiatry 1997; 42:524–526Crossref, Medline, Google Scholar
29 : Central nervous system changes in mitochondrial encephalomyopathy: light and electron microscopic study. Acta Neuropathol 1992; 83:449–452Crossref, Medline, Google Scholar
30 : Juvenile Kearns-Sayre syndrome initially misdiagnosed as a psychosomatic disorder. J Med Genet 1994; 31:45–50Crossref, Medline, Google Scholar
31 : Diabetes mellitus associated with mitochondrial myopathy and schizophrenia: a possible link between diabetes mellitus and schizophrenia. Diabet Med 1997; 14:503Crossref, Medline, Google Scholar
32 : Depressive disorder due to mitochondrial transfer RNALeu(UUR) mutation. Biol Psychiatry 1997; 41:1137–1139Crossref, Medline, Google Scholar
33 : Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes (MELAS) syndrome: an autopsy report. Arch Pathol Lab Med 1998; 122:978–981Medline, Google Scholar
34 : Therapeutic effect of sodium dichloroacetate on visual and auditory hallucinations in a patient with MELAS. Neuropediatrics 1991; 22:166–167Crossref, Medline, Google Scholar
35 : MELAS: a neuropsychological and radiological follow-up study. Mitochondrial encephalomyopathy, lactic acidosis and stroke. Acta Neurol Scand 2002; 106:309–313Crossref, Medline, Google Scholar
36 : MELAS point mutation with unusual clinical presentation. Neuromuscul Disord 1993; 3:191–193Crossref, Medline, Google Scholar
37 : Coenzyme Q10 improves psychiatric symptoms in adult-onset mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes: a case report. Aust N Z J Psychiatry 2000; 34:1034–1035Crossref, Medline, Google Scholar
38 : mtDNA disease in the primary care setting. Arch Intern Med 2001; 161:2497–2500Crossref, Medline, Google Scholar
39 Stewart JB, Naylor, G.J.: Manic-depressive psychosis in a patient with mitochondrial myopathy - a case report. Medical Science Research. 1990 1990;18:2.Google Scholar
40 : Multiple deletions of mitochondrial DNA in several tissues of a patient with severe retarded depression and familial progressive external ophthalmoplegia. J Clin Invest 1992; 90:61–66Crossref, Medline, Google Scholar
41 : Psychiatric disturbance in mitochondrial encephalomyopathy. J Neurol Neurosurg Psychiatry 1989; 52:920–922Crossref, Medline, Google Scholar
42 : Mitochondrial encephalomyopathy (MELAS) with mental disorder. CT, MRI and SPECT findings. Neuroradiology 1990; 32:74–76Crossref, Medline, Google Scholar
43 : Mitochondrial myopathy associated with sudden death in young adults and a novel mutation in the mitochondrial DNA leucine transfer RNA(UUR) gene. Q J Med 1993; 86:709–713Medline, Google Scholar
44 : Psychiatric symptoms in MELAS; a case report. J Neurol Neurosurg Psychiatry 1998; 64:692–693Crossref, Medline, Google Scholar
45 : [A case of mitochondrial encephalomyopathy with schizophrenic psychosis, dementia and neuroleptic malignant syndrome]. Rinsho Shinkeigaku 1991; 31:1219–1223Medline, Google Scholar
46 : Mania as a first presentation in mitochondrial myopathy. Psychiatry Clin Neurosci 2006; 60:774–775Crossref, Medline, Google Scholar
47 : Rare autosomal dominant POLG1 mutation in a family with metabolic strokes, posterior column spinal degeneration, and multi-endocrine disease. J Child Neurol 2010; 25:752–756Crossref, Medline, Google Scholar
48 : Major depression in adolescent children consecutively diagnosed with mitochondrial disorder. J Affect Disord 2009; 114:327–332Crossref, Medline, Google Scholar
49 : POLG1 p.R722H mutation associated with multiple mtDNA deletions and a neurological phenotype. BMC Neurol 2010; 10:29Crossref, Medline, Google Scholar
50 : Autosomal dominant psychiatric disorders and mitochondrial DNA multiple deletions: report of a family. J Affect Disord 2008; 106:173–177Crossref, Medline, Google Scholar
51 : POLG mutations in neurodegenerative disorders with ataxia but no muscle involvement. Neurology 2004; 63:1251–1257Crossref, Medline, Google Scholar
52 : Adult-onset autosomal recessive ataxia with thalamic lesions in a Finnish family. Neurology 2001; 57:1043–1049Crossref, Medline, Google Scholar
53 : Autosomal dominant external ophthalmoplegia and bipolar affective disorder associated with a mutation in the ANT1 gene. Neuromuscul Disord 2003; 13:162–165Crossref, Medline, Google Scholar
54 : Recurrent major depression, ataxia, and cardiomyopathy: association with a novel POLG mutation? Neuropsychiatr Dis Treat 2011; 7:293–296Crossref, Medline, Google Scholar
55 : Mitochondrial transfer RNA(Phe) mutation associated with a progressive neurodegenerative disorder characterized by psychiatric disturbance, dementia, and akinesia-rigidity. Arch Neurol 2010; 67:1399–1402Crossref, Medline, Google Scholar
56 : Multiple neurologic, psychiatric, and endocrine complaints in a young woman: a case discussion and review of the clinical features and management of mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke. Prim Care Companion J Clin Psychiatry 2008; 10:237–244Crossref, Medline, Google Scholar
57 : Psychiatric comorbidity in 36 adults with mitochondrial cytopathies. CNS Spectr 2007; 12:429–438Crossref, Medline, Google Scholar
58 : Depressive behaviour in children diagnosed with a mitochondrial disorder. Mitochondrion 2010; 10:528–533Crossref, Medline, Google Scholar
59 : Treatment-resistant depression. J Clin Psychiatry 2006; 67(Suppl 6):16–22Crossref, Medline, Google Scholar
60 : Vitamin B12, folic acid, and the nervous system. Lancet Neurol 2006; 5:949–960Crossref, Medline, Google Scholar
61 : Extraskeletal effects of vitamin D in older adults: cardiovascular disease, mortality, mood, and cognition. Am J Geriatr Pharmacother 2010; 8:4–33Crossref, Medline, Google Scholar
62 Anglin RE, Rosebush PI, Mazurek MF: Treating psychiatric illness in patients with mitochondrial disorders. Psychosomatics. 2010 Mar;51(2):179; author reply -80.Google Scholar
63 : Neuroleptic medications inhibit complex I of the electron transport chain. Ann Neurol 1993; 33:512–517Crossref, Medline, Google Scholar
64 : Valproate and mitochondria. Biochem Pharmacol 1993; 46:199–204Crossref, Medline, Google Scholar
65 : Fluoxetine interacts with the lipid bilayer of the inner membrane in isolated rat brain mitochondria, inhibiting electron transport and F1F0-ATPase activity. Mol Cell Biochem 1999; 199:103–109Crossref, Medline, Google Scholar
66 : The effects of antidepressants on mitochondrial function in a model cell system and isolated mitochondria. Neurochem Res 2011; 36:327–338Crossref, Medline, Google Scholar
67 : Mitochondrial diseases in man and mouse. Science 1999; 283:1482–1488Crossref, Medline, Google Scholar
68 : A neurological perspective on mitochondrial disease. Lancet Neurol 2010; 9:829–840Crossref, Medline, Google Scholar

