
Role of Serotonin in the Behavioral and Psychological Symptoms of Dementia
Abstract
The behavioral and psychological symptoms of dementia (BPSD) can have serious debilitating effects on the patient and increase caregiver burden. Investigations into the underlying neuropathology indicate that the serotonergic system may contribute to BPSD. In addition, serotonergic pathways are known to interact extensively with the cholinergic, noradrenergic, GABAergic, and dopaminergic systems. Hence, serotonergic therapies may be used to manipulate other neurotransmitters systems to alleviate BPSD or in combination with agents specific for the other neurotransmitter receptor sites. Neurotransmitter-modulated behaviors and evidence provided by pharmacological interventions are reviewed, focusing primarily on the serotonergic system.
Behavioral disorders have long been recognized as a common feature in Alzheimer's disease (AD) and related dementias. The nature and frequency of the behavioral and psychological symptoms of dementia (BPSD) have been outlined in numerous papers.1,2 BPSD can include psychotic symptoms such as hallucinations and paranoid/delusional ideation, nonpsychotic symptoms such as aggression and wandering, and affective disturbances and anxieties/phobias.1 BPSD occur throughout the course of dementia, irrespective of level of cognitive impairment.3 It has been estimated that up to 90% of patients display BPSD.1,4 Frequently identified BPSD include delusions (10% to 73%), wandering (10% to 70%), aggression (18% to 65%), motor restlessness (21% to 60%), hallucinations (3% to 49%), and depression (30% to 40%).1,2,5 These BPSD are distressing to the caregivers and are factors stimulating the decision to institutionalize.5,6 Furthermore, the presence of BPSD may have a negative impact on the course of the disease.7–9 Knowledge of the importance of BPSD for both caregivers and patients has stimulated research into their underlying pathological and neurochemical substrates.
Strong, consistent evidence supports the presence of significant disruptions in global serotonergic neurotransmission in dementia.10 Serotonergic neurons originating from the dorsal and median raphe nuclei innervate many structures in the cortex and limbic system.11,12 These widespread projections enable the serotonergic system to regulate aggression, mood, feeding, sleep, temperature, sexual activity, and motor activity.11,13 Therefore, alterations in the functioning of the central serotonergic system can be expected to have a clinically discernible impact on behavior.
The serotonergic system is heterogeneous in terms of structure and function, making it a complex system to study. Moreover, the dysfunction of the serotonergic system studied in isolation cannot be expected to explain changes in behavior, given that many neurotransmitters work in conjunction with each other. For example, although serotonin (5-hydroxytryptamine; 5-HT) generally plays an inhibitory role in the human cortex, overall excitability is mediated by acetylcholine, γ-aminobutyric acid (GABA), noradrenaline, histamine, and purines.14 There is some evidence that each of these neurotransmitters is altered in AD and related dementias,15–20 and each has a role in controlling human behavior. Hence, optimum treatment efficacy may be achieved by correcting multiple neurotransmitters or the balance between neurotransmitters.
We will review evidence supporting neurochemical alterations and putative links with BPSD for the serotonergic system and four neurotransmitters that interact extensively with serotonin: acetylcholine, noradrenaline, dopamine, and GABA. For each section, the functional role of the neurotransmitter, the evidence supporting a disruption in AD, and any studies indicating a link with BPSD will be reviewed. Any evidence for other dementias will immediately follow. Neurotrophic factors also control behavior. The neuropeptides somatostatin, neuropeptide Y, and brain-derived neurotrophic factor also interact extensively with serotonin and will be briefly discussed.
Articles were retrieved from various sources, including electronic databases (MEDLINE, EMBASE) and cross-references from relevant articles. The following keywords were used: serotonin, acetylcholine, dopamine, norepinephrine, gamma-aminobutyric acid, GABA, neuropeptides, neurotrophic factors, neuropeptide Y, somatostatin, brain derived neurotrophic factor, Alzheimer's disease, dementia, behavior, behavioral disorders, delusions, hallucinations, agitation, aggression, apathy, disinhibition, depression, cognition, and non-cognitive.
SEROTONERGIC SYSTEM
The evidence supporting the role of serotonin disruption contributing to BPSD consists of biologic plausibility, demonstration of disruptions in dementia, studies linking disruptions with BPSD, and clinical evidence from serotonergic medications. Most of the studies have been done in patients with Alzheimer's disease (Figure 1). This evidence is reviewed below.
In Vitro Evidence of Serotonergic Disruption in Dementia
Neurochemical and neuropathological disruptions in the serotonergic system have been established in AD. Loss of neurons in the raphe nuclei has been demonstrated,21,22 as has loss of serotonergic nerve terminals in the neocortex.15 Decreased concentrations of 5-HT and its major metabolite 5-hydroxyindoleacetic acid (5-HIAA) have been demonstrated in the central nervous system by use of postmortem brain studies, particularly in the temporal cortex16,17,19,23–27 and in the cerebrospinal fluid (CSF).28 Although indirect, taken as a whole these data provide evidence that there are changes in the actual functional status of central serotonergic transmission. The presence of expected adaptive consequences due to serotonergic lesions, such as supersensitivity, tolerance, trophic changes, or other changes in 5-HT-modulated central functions,29 remains to be elucidated.
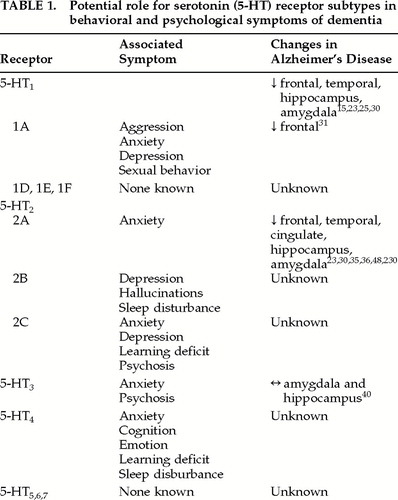
The actions of 5-HT are mediated through at least seven major receptor classes that have differing placements in the synapse, utilize different second messenger systems, and have different locations in the brain.12,29 5-HT1A receptors are located both presynaptically (somatodendritic in the raphe nuclei) and postsynaptically (terminals in the limbic system, hypothalamus, hippocampus, frontal cortex, and brainstem) and can have contradictory actions on intracellular signaling.12,29 This receptor subtype is thought to be involved with anxiety, depression, sexual behavior, and aggression12 (Table 1), as well as appetite. The functions influenced by 5-HT1Dα, 5-HT1Dβ, 5-HT1E, and 5-HT1F are still being elucidated, and functions identified so far are not related to behavior.12 Deficits involving 5-HT1 receptors in cortex, hippocampus, and amygdala have been shown to exist in AD by ligand binding studies.15,23,25,30–32 However, loss of 5-HT1 receptors is also age-related in people with AD.23 The 5-HT1A selective agonist 8-hydroxy-2-(di-n-propylamino) tetralin (abbreviated 8-OH-DPAT) has been used to suggest that 5-HT1 decreases are in the 5-HT1A receptor subtype.31,32 Given the actions of 5-HT1A, alterations in this subtype may be highly relevant to BPSD in AD.
There are three subtypes of 5-HT2 receptors, all of which have some involvement in behaviors (Table 1). 5-HT2A is located in the cortex and caudate and is involved in anxiety (as well as vasoconstriction and migraine); 5-HT2B is a postsynaptic receptor found in the limbic system and hypothalamus that has been associated with depression, sleep, and hallucinations; and 5-HT2C is a postsynaptic receptor that has been associated with anxiety, depression, learning, and psychosis (as well as appetite and aversion).12 Deficits involving 5-HT2 receptors in AD frontal and temporal cortex, cingulate, hippocampus, and amygdala have been demonstrated with ligand binding studies,23,30,33,34 autoradiographic studies,35 and positron emission tomography (PET) studies.36 Loss of 5-HT2 receptors is much greater than loss of 5-HT1 receptors in AD23,34,37 and is not seen in other dementing illnesses.11,34 One study, which found 5-HT2 receptor binding was unchanged in the frontal cortex and hippocampus, may have been confounded by use of psychotropic medications at the time of death.38 Given the magnitude of the deficit in 5-HT2 receptors and the involvement of these receptors in behavior, this serotonergic system may also be important in BPSD associated with AD. Polymorphic variations have been identified for 5-HT2A and 5-HT2B receptors that may be risk factors for BPSD such as visual hallucinations.39
Relationships between the remaining 5-HT receptors and behavioral disorders seen in AD are not as well supported. The 5-HT3 receptor is postsynaptic and found in the cortex, midbrain, limbic system, hippocampus, and vomiting centers.12 It has very little homology with other 5-HT receptors and uses a difference second messenger system.12,29 Behaviors modulated by 5-HT3 are thought to include anxiety and psychosis,12 and thus this receptor subtype has a potential role to play. However, loss of 5-HT3 receptors was not demonstrated in the amygdala and hippocampus of AD patients compared with age-matched control subjects.40 The 5-HT4 receptor is found postsynaptically in the hippocampus, frontal cortex, limbic system, superior colliculi, and basal ganglia and has been implicated in learning, cognition, emotion, anxiety and sleep.12 Alterations in this subtype have not yet been identified in AD. The functions and locations of 5-HT5, 5-HT6, and 5-HT7 remain unclear, and their dysregulation in AD and related dementia has not been studied (Table 1). Receptor-specific ligands are currently being developed for 5-HT5,6,7.29
Preliminary studies show that AD may also be associated with changes in 5-HT signal transduction. Alterations in 5-HT1A receptor coupling to guanine nucleotide binding (G) proteins have been demonstrated in the superior frontal cortex,41 although another group found coupling to be preserved in the cerebral cortex.42 Uncoupling of phosphoinositide (PPI) hydrolysis from 5-HT stimulation in frontal cortex tissue of AD brains compared with age-matched controls has also been demonstrated.43 Study of second messenger systems is important because uncoupling implies that neurotransmitter replacement strategies would be ineffective.
Disruptions in serotonergic neurotransmission have also been studied in other dementing illnesses. Serotonin binding was reduced by 50% in the putamen of patients with the multi-infarct type of vascular dementia (VaD).44 Serotonin deficits were also found in a non– multi-infarct category of VaD in cortical and subcortical gray matter.45 However, a more recent study was unable to demonstrate any serotonin deficits.46 Radioligand binding showed an intact brain serotonin system both presynaptically (uptake with [3H]paroxetine) and postsynaptically (5-HT1A with [3H]8-OH-DPAT and 5-HT2 with [3H]ketanserin) in the frontal cortex, temporal cortex, and caudate nucleus (presynaptic only) in vascular dementia.46 As expected, serotonin deficits may depend upon the subtype of VaD and the extent and location of lesions. There is also preliminary evidence for serotonergic changes in Pick's disease47 and Lewy body dementia.48,49
In Vivo Studies Examining Linkage With BPSD
Neuroendocrine studies have been performed in AD patients that support the role of 5-HT in behavior control. In one study using the serotonin agonist m-chlorophenylpiperazine (m-CPP), patients with AD (n=12) showed increased behavioral responsivity (psychomotor activation, restlessness, and perceptual abnormalities) compared with 10 age-matched control subjects.50,51 This may reflect damage to the serotonergic system with subsequent up-regulation of the remaining postsynaptic receptors.11 Unexpectedly, the neuroendocrine response to m-CPP did not differ between the two groups. However, since m-CPP binds to 5-HT1A, 5-HT2, and 5-HT3, as well as α1-, α2-, and β-adrenergic sites, dopaminergic and cholinergic sites, discrepancies between the neuroendocrine response and the behavioral response of m-CPP may be due to actions on nonserotonergic neuronal systems. Although this study indicates a possible link between 5-HT and behavior, unfortunately there was no assessment for presence of BPSD in these patients. McLoughlin et al.52 performed a neuroendocrine study that confirmed the presence of a 5-HT hyperresponsivity in AD. Unfortunately, no attempt was made to relate this to the presence or absence of behavioral disturbances.
The relationship between decreased serotonin and specific behaviors in patients with AD and other dementing disorders has been studied (Table 2). Postmortem studies have found decreased 5-HT levels in AD patients with a history of psychotic behaviors, compared with nonpsychotic AD patients, in some areas of the brain.53 However, another study looking at the temporal cortex and both psychotic and nonpsychotic symptoms in patients with AD was unable to confirm these findings.54 Chen et al.26 found that concentrations of serotonin in the frontal cortex and 5-HIAA in the temporal cortex were significantly lower in patients on chronic neuroleptic treatment compared with patients not receiving neuroleptics. Because previous studies did not control for use of neuroleptics, it is unclear whether decreased serotonin is related to psychotic behaviors or to treatment with neuroleptics in AD. In Lewy body dementia, there is some evidence that preservation of 5-HT2 receptors in the temporal cortex may be associated with hallucinations.48
Serotonin has also been related to nonpsychotic BPSD in both postmortem and clinical studies. A clinical study using CSF levels of the 5-HT metabolite 5-HIAA found that levels of 5-HIAA in the CSF were positively correlated with anxiety and fear/panic.55 Decreased cortical levels of 5-HT were found post mortem in patients with a history of aggression compared with nonagitated patients,19 whereas normal numbers of 5-HT2 receptors were found in nonaggressive patients.56 Clinical studies looking at putative peripheral markers of serotonergic activity have also been performed. Schneider et al.57 evaluated the relationship between binding to the platelet serotonin transporter system (a marker of serotonergic activity) and BPSD in patients with AD. The BPSD were predominantly agitation and delusions. The agitated/delusional groups showed significantly lower peripheral serotonergic activity than AD subjects without BPSD.57 However, a previous report by Suranyi-Cadotte et al.58 found no differences in the density of platelet serotonin binding sites between 9 AD patients and 11 age-appropriate controls when subjects with depressive symptoms had been excluded. Although that study58 demonstrated a link with depression, a recent postmortem study was unable to confirm this link.27 Because depressed patients were not explicitly excluded in the Schneider et al. study57 and the nonagitated group had apathy, it is unclear whether the association with agitation and delusions can be partially or wholly attributed to comorbid depression in some of these patients. Furthermore, platelet 5-HT2 function may not be adequate as a peripheral marker for the central serotonergic system. For example, a negative correlation has been found between CSF 5-HIAA and receptor indexes in other neuropsychiatric patients.59
Recently, two neuroendocrine studies found that agitated aggressive patients had an increased response to the 5-HT releasing agent fenfluramine compared with nonagitated aggressive patients.60,61 Both neuroendocrine BPSD studies excluded AD patients with significant depression. One study also found a significant gender effect.62 Gender has not been controlled for in other studies looking at the serotonergic system and may be an important contributor to variations in the serotonergic system throughout the life span.
In summary, in vitro and in vivo studies provide inconsistent evidence to link serotonin dysfunction with psychotic symptoms and with anxiety and depression in AD. Failure to control for concomitant medication use and comorbidity are largely responsible for these inconsistencies. Current clinical evidence does support earlier work by Palmer et al.19 and Procter et al.56 linking aggressive agitation with the serotonergic system.
Evidence From Pharmacotherapy Studies
The neurochemical and neurophysiological evidence that the serotonergic system may contribute to BPSD has been supported by a number of studies involving pharmacologic agents specific for the serotonergic system. Clinical manipulation of the serotonergic system through pharmacotherapy provides only indirect evidence of the involvement of that system with the target behavior. However, because the goal of understanding BPSD is to improve treatment, this information is worth briefly reviewing. (More extensive reviews are available.1,63) Considering there are 15 different serotonergic receptor subtypes, the number of serotonergic selective medications is limited. Agents reviewed here include the selective serotonin reuptake inhibitors (SSRIs), atypical antipsychotics, buspirone, ondansetron, and the serotonin and noradrenaline reuptake inhibitors (SNRIs).
The SSRIs alaproclate,64 zimeldine,65 fluoxetine,66,67 citalopram,68 fluvoxamine,69 paroxetine,70 and sertraline71 have been given to patients with BPSD in randomized controlled trials. Many patients have also been treated with open-label SSRIs, with positive results. For example, in an open-label study by Pollock et al.,72 9 of 16 patients receiving citalopram showed significant improvement in disinhibition, agitation, hostility, and suspicion. However, when SSRIs have been administered under double-blind, placebo-controlled conditions, results have been equivocal with respect to efficacy for BPSD. Randomized controlled trials in AD patients with alaproclate64 and fluoxetine66 have shown little efficacy for the SSRIs in treating BPSD, except for depression.67 Placebo-controlled trials with mixed dementia populations have shown nonsignificant improvement in BPSD such as irritability, anxiety, fear/panic, mood, and restlessness with fluvoxamine,69 and significant improvements in depression with paroxetine.70 Fluoxetine was reported to slightly improve disinhibition, depressive symptoms, carbohydrate craving, and compulsions in about one-half of 11 patients with frontotemporal dementia.73 The strongest efficacy was demonstrated by citalopram, which, among the SSRIs, has the greatest in vitro selectivity ratio for the serotonergic versus the noradrenergic system.74 Nyth and Gottfries68 administered citalopram for 4 weeks to patients with mild to moderate AD (n=65) or multi-infarct dementia (n=24) in a multicenter, placebo-controlled, parallel group study. In AD patients, significant improvements were noted on a geriatric rating scale in emotional bluntness and in all six BPSD (restlessness, confusion, irritability, anxiety, fear/panic, and depressed mood) for baseline versus citalopram scores. Irritability and depression improved significantly for citalopram versus placebo groups. There was no significant treatment effect in the multi-infarct group. The patients in this study had very mild BPSD at baseline, with mean scores of less than 2 out of 6 on each symptom.
The atypical antipsychotics (e.g., risperidone, clozapine, olanzapine) are being used more frequently in patients with BPSD because of their efficacy in treating psychotic symptoms and their favorable side effect profiles compared with their typical antipsychotic counterparts (i.e., reduced extrapyramidal effects). Most of the new antipsychotics have strong antagonistic affinity for the 5-HT2 receptor. Because the hyperresponsivity of the serotonergic system noted in agitated and aggressive AD patients60,61 suggests up-regulation of 5-HT2 receptors, the atypicals may be of benefit in the management of BPSD. To date, risperidone, clozapine, olanzapine, and quetiapine have been assessed to treat behaviors related to dementia.
Risperidone, in a total of 534 patients from case reports,75–81 case series,82,83 retrospective chart reviews,84–87 and open-label cohort studies,88–90 was reported to produce significant reductions in behaviors such as psychosis and agitation in patients with various types of dementia. Further evidence of risperidone efficacy for BPSD was reported in two recent randomized controlled trials. Katz et al.91 reported on 625 patients, diagnosed with AD and/or vascular dementia, who received either placebo or 0.5 mg, 1 mg, or 2 mg of risperidone for 12 weeks. The authors found a dose– response relationship with risperidone for reducing all behaviors and aggressiveness, and psychotic symptoms were significantly reduced with 1 mg, but not 2 mg, of risperidone compared with placebo. De Deyn et al.,92 in a 12-week trial of patients with AD and/or vascular dementia, found risperidone to significantly reduce aggressiveness compared with haloperidol and placebo.
Clozapine has not been evaluated for the treatment of BPSD in any randomized controlled trials. In an open-label dechallenge and rechallenge case series by Oberholzer et al.,93 clozapine was found to significantly improve “antisocial behavior,” “social competence,” “social interest,” and “irritability” in 7 patients with AD. In two retrospective chart reviews,94,95 clozapine (doses of 100–300 mg per day) was reported to have minimal to moderate effects on behaviors (e.g., psychosis, agitation) in 5 patients with a variety of dementia diagnoses. Frankenburg et al.96 reported a marked response (reduced hallucinations and verbal aggression) to low-dose clozapine (i.e., 18.5 mg/day) in a patient with parkinsonian dementia. However, 2 other patients with unspecified dementia were reported to have no changes or minimal change in behaviors with clozapine.96 Finally, in a case report of 4 patients at varied stages of dementia, Pitner et al.97 reported marked improvement in psychosis and other behaviors in 2 patients, and worsening of behavioral symptomatology in the remaining 2 patients.
In a large open-label trial of olanzapine, Kinon et al.98 reported that patients diagnosed with AD and/or vascular dementia showed significant improvements in psychosis and/or agitation. A recent randomized controlled trial by Street et al.99 reported olanzapine (5 mg and 10 mg doses) to significantly improve symptoms of psychosis and other behaviors compared with placebo in 206 AD patients over a 6-week period. The authors also reported that fewer patients without psychosis treated with olanzapine developed delusions and/or hallucinations by the end of the study compared with those receiving a placebo. Walker et al.100 reported that 6 of 8 patients diagnosed with Lewy body dementia responded to open-label olanzapine with significant reductions in BPSD.
Quetiapine, in a single published open-label study by McManus et al.,101 was found to significantly improve overall BPSD and psychotic symptoms in 106 patients with a variety of organic diseases over a 12-week period.
Although the atypical antipsychotics appear to improve BPSD, it is unclear whether this result is secondary to serotonergic or dopaminergic antagonism, since the typical antipsychotics (e.g., haloperidol, perphenazine, thioridazine) have also been demonstrated to have significant benefit.102 Future research might help to determine if typical and atypical antipsychotics have different patterns of symptom improvement.
Buspirone (a 5-HT1A partial agonist), in two randomized controlled double-blind trials, was compared with placebo10 and haloperidol103 to determine efficacy in treating BPSD. There was no significant overall reduction in behavioral symptoms with buspirone compared with placebo in the small number of patients tested (N=10). However, in the other study, buspirone was as effective as haloperidol in treating 26 patients with probable AD and superior to haloperidol in decreasing anxiety and tension.103 This latter study did not have a placebo control. Thus, the evidence for the efficacy of buspirone for BPSD is not strong.
Ondansetron (a 5-HT3 selective antagonist), used primarily for the management of chemotherapy-induced nausea and vomiting, has been used in patients with Parkinson's disease accompanied by dementia.104,105 Both open-label trials showed some benefit in alleviating visual hallucinations and delusions. Further evidence linking ondansetron to a reduction in behavioral problems awaits controlled clinical trials.
The SNRIs trazodone and clomipramine have also been tested for BPSD. Trazodone, in a number of open-label studies, was found to improve behavioral symptoms in 80% of demented elderly patients enrolled.106–112 In a double-blind trial, trazodone showed a small but significant reduction in behavioral symptoms compared with buspirone and placebo.10 In another double-blind study controlled with haloperidol, trends suggested that specific symptoms, including aggression, may respond preferentially to trazodone.113 Clomipramine, another SNRI, produced significant overall improvement in depression in a small double-blind crossover study of AD patients.114 Although evidence to date shows some benefit with SNRIs, these drugs have more affinity for the noradrenaline than the serotonergic receptors and cannot be considered serotonin-selective medications.
In summary, the evidence supporting the efficacy of SSRIs, atypical antipsychotics, 5-HT agonists and antagonists, and SNRIs in the management of BPSD is as yet inconclusive. However, it must be noted that, as with other trials for BPSD,102 methodological issues with these studies included failure to blind or control,71–73,75–90,93–98,100,101,106–111,115–130 insufficient power,10,66,73,94–97,100,101,106–111,113 inclusion of a heterogeneous population (patients had varying etiologies for their dementias or were at varying stages of the illness or had different subtypes of BPSD),64,68–70,77–79,81–83,85,86,88,89,91–98,113,117,129,130 and failure to use standardized and validated scales to identify and track target behaviors.64,66,77,78,82,85,86,88,94–96,103,113–115,119,120,122–124,130 Furthermore, improper dosing and duration of treatment could contribute to some negative studies, since the effective dose and duration for treatment of different BPSD are unknown. Thus far, there is some evidence to support the use of serotonergic medications. The preliminary literature on SSRIs supports the use of the most serotonin-selective of these medications in patients with Alzheimer-type dementia, and this is corroborated by positive results with atypical antipsychotics. However, there are no predictors of response.
OTHER NEUROTRANSMITTER SYSTEMS
Acetylcholine (ACh)
Loss of cholinergic neurons is an early and consistent finding in AD and is thought to be essential to the pathophysiology. Numerous studies have demonstrated profound changes in the cholinergic system in AD, including deficits in the major cholinergic system arising in the basal forebrain and projecting to the cortex;131–134 decreases in the cholinergic markers choline acetyltransferase (ChAT) and acetylcholinesterase (AChE) in the cortex, particularly the temporal cortex;24,38,135–137 significant losses of neurons in the nucleus basalis of Meynert;131,138 and reductions in the muscarinic type 2 (M2) presynaptic receptor density.132,137 Cholinergic neurons arise in the basal forebrain (nucleus basalis of Meynert, diagonal band of Broca, and medial septum) and innervate widespread areas of the cortex, including the hippocampus and a variety of subcortical structures. Therefore, alterations in the central cholinergic system can be expected to cause significant disruptions in the functioning of the neocortex.
Although the role of the central cholinergic system in cognition is well recognized,139–145 there is only preliminary evidence suggesting that this neurotransmitter plays an important role in the noncognitive disorders associated with dementia. The cholinergic hypothesis of noncognitive symptoms of AD put forth by Cummings and Back146 suggests that cholinergic deficits may contribute to symptoms such as psychosis-agitation, apathy-indifference, disinhibition, and aberrant motor behavior. Since virtually all AD patients have loss of cholinergic neurons but some do not have BPSD, other neurotransmitters may be involved.
Serotonin and acetylcholine interact extensively in the human brain. For example, 5-HT inhibits release of ACh from cortical and hippocampal cholinergic nerve terminals, possibly via 5-HT1B receptors in the hippocampus.11 The 5-HT3 receptors may also inhibit the release of ACh, whereas 5-HT1A receptors may mediate an increase in ACh release.29 Thus, disruptions in 5-HT have the potential to influence an already compensated cholinergic system.
A single postmortem study by Procter et al.56 examined 5-HT receptor binding (with [3H]ketanserin) and ChAT activity in 17 dementia patients with or without aggressive symptomatology (4 aggressive patients, 13 nonaggressive patients) compared with 18 healthy elderly control subjects. Except for the superior parietal lobe, [3H]ketanserin binding was decreased in 12 cortical areas in aggressive compared with nonaggressive patients. However, ChAT activity throughout the cortex was similar in the two patient groups and was significantly lower in these groups compared with healthy control subjects. Thus, current evidence supports 5-HT deficits being more closely related than acetylcholine to aggression in AD.
Clinical experience with cholinergic and anticholinergic medications provides an indirect link between BPSD and the cholinergic system. In AD, cholinergic therapies have been reported to affect psychosis and agitation, apathy/indifference, disinhibition, and aberrant motor behavior.146 Postmortem studies provide some evidence that neuronal losses in the nucleus basalis, and also decreases in cholinergic transmission, are correlated with psychotic symptoms in Lewy body dementia.147,148 Choline acetyltransferase was also shown to be decreased in Lewy body dementia patients with hallucinations.49 Overall, there is some preliminary evidence linking psychotic behavioral changes with the cholinergic system, especially the balance between this system and the monoamines.49 In summary, evidence supports the suggestion that the addition of serotonergic and cholinergic deficits may be responsible for aggression and psychotic symptoms.
The question of whether serotonergic changes are secondary to cholinergic changes or independent of them cannot be answered as yet. To date, there are no longitudinal studies identifying parallel progression in the degeneration of the 5-HT and cholinergic systems. There are some studies that have identified concurrent deficits25,149–152 or no change15,153,154 in 5-HT (e.g., 5-HT and 5-HIAA concentrations, [3H]5-HT binding) with respect to loss of cholinergic markers (e.g., ChAT, ACh synthesis) in AD patients compared with control subjects. These findings suggest that serotonergic deficits are either a variable component of AD or occur only in advanced AD.
Noradrenaline (NA)
The central serotonergic and noradrenergic systems interact in many areas. Serotonin is a co-transmitter with NA, and uptake of 5-HT and NA can be accomplished by either 5-HT or NA neurons.155 The serotonergic system also inhibits the release of NA via 5-HT1 receptors.29 Thus, there is reason to suspect that dysfunction in the serotonergic system will be accompanied by changes in the NA system. Animal studies have shown that NA neurons from the locus ceruleus (LC) are involved in behaviors such as the sleep-wake cycle, level of vigilance, and emotion.156,157 Dysfunction in the NA system may also directly influence BPSD.
Postmortem studies suggest AD is associated with loss of neurons in the locus ceruleus,134,158 the major source of NA in the brain, and with decreased levels of NA in many brain areas innervated by the LC.17,20,24,27,53,159,160 Loss of NA neurons is correlated with the severity of dementia.161 In contrast, levels of the NA metabolite 4-methoxy-5-hydroxy-phenylglycol (MHPG) are increased,162–165 indicating that NA metabolism is increased. This increased MHPG/NA ratio likely reflects compensatory increases in the activity of remaining LC neurons.27 These changes are not reflected in the CSF of patients.160,166 CSF levels of MHPG are thought to increase with advancing disease,167,168 complicating interpretation of CSF studies.
Evidence supports a link between depressive symptoms in AD patients and NA dysfunction. The evidence includes neuronal losses in the LC,156,169–171 lower levels of cortical NA,172 and increased NA dysfunction.173 However, a well-designed postmortem study with prospective patient evaluations before death was unable to confirm this relationship.27
Preservation of NA may also be associated with BPSD. A postmortem study in AD patients indicated that increased levels of NA,53 comparable to those found in elderly control subjects,172 were associated with psychotic behaviors, but this relationship was not reflected in CSF MHPG concentrations.168 There is evidence that restlessness is weakly related to CSF levels of MHPG in patients with AD,55 although agitation, in another study, was not.168 A recent study also showed markedly increased concentrations of α2 receptors (by ∼70%) and β1 and β2 adrenergic receptors (by ∼25%) in the cerebellar cortex of aggressive patients compared with nonaggressive patients with similar levels of cognitive deficit. Levels of α2 receptors in aggressive patients were slightly above those in the healthy elderly control subjects, whereas increases in β receptors were higher than in both nonaggressive AD patients and control subjects.174 Thus, aggressive behavior may be associated with cerebellar noradrenergic preservation.
Dopamine (DA)
Serotonergic neurons interact closely with dopaminergic neurons. For example, serotonergic neurons from the raphe nuclei are known to synapse with dopaminergic neurons and to control dopamine release in the midbrain, striatum, and nucleus accumbens.175 Serotonergic neurons may either inhibit (via 5-HT1A) or increase release of DA.29 Thus, loss of serotonergic neurons will affect the DA system.
The dopaminergic system has been implicated in depression, agitation, and psychotic behaviors in nondemented patients, and thus this system has the potential to directly influence BPSD. Although disruptions in the dopaminergic system have been demonstrated post mortem in AD patients compared with control subjects,20,159 DA is less affected than other biogenic amines.17,27 Dopaminergic disruptions may primarily involve presynaptic disruptions in the DA transporter and postsynaptic loss of striatal dopamine type 2 (D2) receptors.176 Furthermore, loss of striatal D2 receptors may be more common in the 30% of AD patients who also show extrapyramidal features.176,177 Interestingly, a neuroimaging study demonstrated that disruptions in DA metabolism became increasingly severe as the cognitive impairment progressed.178 As a result, level of cognitive impairment will be a confounder for any studies attempting to relate DA dysfunction to behavior.
Studies in patients with AD that controlled for level of cognitive impairment have not been able to demonstrate the expected links between psychotic behaviors and abnormalities in the DA system53,179,180 or depression and the DA system.27 The same may not be true in other dementias. In Lewy body dementia, dopamine metabolites were significantly decreased in nonhallucinating patients in conjunction with serotonergic abnormalities (i.e., decreased serotonergic 5-HT2 receptor binding and decreased 5-HT metabolites).49 When combined with the finding that ChAT was decreased in hallucinating patients, these results suggest that an imbalance between monoaminergic and cholinergic transmitters is involved in hallucinations seen in Lewy body dementia.
There is some evidence that agitated and aggressive behaviors may be associated with a relative preservation of DA function in AD patients. A postmortem study181 demonstrated that preservation of substantia nigra neurons was related to aggression and interpersonal violence. Studies performed in vivo have supported this. Lopez et al.182 found that whereas aggressive patients showed similar CSF homovanillic acid (HVA) concentrations to their age-matched controls, nonaggressive patients showed decreased levels. Similarly, Sweet et al.183 found that improvement in hostility during perphenazine treatment correlated with change in plasma HVA levels in 21 patients (89% AD), further supporting the link between aggression and DA. Thus, DA preservation may be associated with the presence of aggressive behaviors. It is plausible that cholinergic and serotonergic dysfunction may also be present and necessary for this behavior. There are no studies answering this question as yet.
Gamma-aminobutyric Acid (GABA)
Serotonin is a co-transmitter with GABA, and GABA agonists can alter the function of several 5-HT receptors.184 GABA is the primary inhibitory neurotransmitter in the CNS.185 It is a local inhibitory interneuron for other neurotransmitters that are key in controlling behavior, including serotonin and dopamine.186 In addition to these local circuit actions, GABAergic neurons project from the striatum to the lateral globus pallidus and from the cerebellar inferior olivary nucleus to the vestibular nucleus.186 Thus, GABA is well positioned to influence many psychobiological functions such as behavior through interactions with serotonin.
Involvement of the neurotransmitter GABA has been shown in behaviors such as aggression, increased GABA being associated with decreased aggression.187 Many lines of evidence have been pursued to establish the presence or absence of a GABAergic abnormality in AD, including postmortem studies of neurotransmitter concentrations150,188–198 and binding,18,35,37,199–207 antemortem studies of CSF levels of neurotransmitter and metabolites,208–216 and PET217,218 and SPECT219 studies. Each of these avenues has generated inconclusive results; many studies showed decreased GABA,18,37,189–195,197,199–201,203,205,206,209,210,215 but some studies showed no significant changes.150,188,196,207,208,211–214,216 Although the discrepant results could reflect the varying methodologies used, each with inherent limitations, these results may also reflect reality. This evidence suggests that GABA disruption is not linked to cognitive degeneration to the same extent as cholinergic dysfunction. GABA dysfunction is a variable component of AD, and, as such, may be linked to the presence of certain behavioral disorders. As yet, there are no clinical studies linking changes in the GABA system with particular behaviors seen in AD.
Neuropeptides
Neurotrophic factors, such as brain-derived neurotrophic factor, somatostatin, and neuropeptide Y, have all been reported to be co-localized within serotonergic neurons in the CNS in both animals and humans.220–222 Thus, loss of serotonergic neurons implies at least some disruption in these neuropeptides. Although a number of authors have studied the relationship between neuropeptide abnormalities and AD pathology (see review by Gottfries et al.223), very little work has focused on the putative effects of altered neuropeptides and BPSD. Minthon and colleagues, in two separate studies,224,225 attempted to correlate somatostatin and neuropeptide Y levels in the CSF with BPSD. The authors reported a significant negative correlation of neuropeptide Y levels with clinical features of AD, including anxiousness, restlessness-agitation, and irritability. The authors failed to find a significant correlation between somatostatin and specific behaviors related to dementia. Whether a reduction in neuropeptide Y correlates with serotonergic deficits in patients with BPSD has yet to be determined. However, given that many of these neurotrophic factors have been associated with behaviors (e.g., aggression, anxiety, depression) in nondemented populations,226 it is plausible that reductions in specific neuropeptides concurrently with serotonin can contribute behavioral phenotypes in dementia.
CONCLUSIONS
The serotonergic system is complex, and much remains to be learned about its function in normal states and dementia. Current evidence suggests that AD and related dementias are associated with changes in serotonergic neurotransmission. In addition, many neurotransmitters and neuropeptides that interact extensively with the serotonergic system are dysfunctional in these dementias, but it is unknown whether this is secondary to cholinergic or serotonergic changes. Attempts to link these changes with specific behavioral and psychological symptoms of dementia have been made. Although there is no clear evidence linking alterations of the serotonergic system with a single behavior or cluster of behaviors, it seems relatively certain that such alterations do contribute to noncognitive symptoms. The most supportive evidence is for a contribution of serotonergic system changes to aggressive behaviors and psychotic behaviors.
Much of the evidence to date consists of postmortem studies. For these studies to be rigorous, it is necessary to have prospective, antemortem data collection, including data on drug use and on well-defined behaviors. Neuroimaging techniques (PET, SPECT), for example, represent novel approaches to determining neurotransmitter changes in antemortem brain. Radiolabeled agonists or antagonists specific for each receptor system can help to elucidate altered binding potential of certain receptor subtypes. For example, the 5-HT1A–specific antagonist [11C]WAY-100635 has been used successfully to determine 5-HT1A binding potential in patients with depression.227,228 In the case of dementia, these radiolabeled tracers may lead to the identification of specific receptor deficits that may correlate with noncognitive aspects of the disease—which in turn may lead to receptor-specific drug therapies. Other potential antemortem investigations of central neurotransmitter function include neuroendocrine challenge studies to predict response to drug treatment (e.g., prolactin response to serotonergic challenge, as mentioned previously), prospective intervention studies of multiple drug therapies to manage the spectrum of behaviors (e.g., neuroleptics and antidepressants), and longitudinal studies with drug prophylaxis in early dementia (e.g., SSRI administration, combined with longitudinal scanning, in dementia patients with a family history of mood disorders).
Unfortunately, much of the work done to date suffers from lack of standardized definitions for behaviors. Patients were frequently defined clinically by the presence of a given behavior. Because many of the behaviors overlap, selecting on the basis of the presence of a single behavior will not provide homogeneity in the patient population. A neurotransmitter-based approach will offer information that can be more closely linked with treatment options. Another limitation is that in postmortem studies linking behaviors to neurotransmitter abnormalities, the behaviors may or may not be expressed at the time of death. Because behavioral changes associated with degenerative neurologic changes are likely to be predominantly a state rather than a trait, it is important that the behaviors are expressed near the time of death. Also, both neurotransmitter disruptions and behaviors are known to change with level of cognitive impairment.229 Findings of changes in behaviors may arise both from actual changes in the behaviors and difficulty in measuring some behaviors as the level of cognitive impairment increases (e.g., auditory hallucinations as the patient becomes less verbal). Thus, it is important to control for level of cognitive impairment as well as to study BPSD through the course of the disease. This latter goal will be more easily achieved with the application of in vivo measures of receptor function; these can include peripheral markers such as neuroendocrine challenges and CSF levels, as well as functional imaging when feasible.
Studying the neurochemical etiology of BPSD presents many challenges. The activity of a single neurotransmitter such as serotonin cannot be separated from those of other neurotransmitters and neuropeptides. Hence, it is essential that future work focus on identifying concurrent neurotransmitter and neuropeptide changes through the progression of the disease. Careful study of clusters of BPSD and their corresponding imbalances may eventually provide enough information to make possible evidence-based, patient-specific treatments for behavioral disorders in dementia.
ACKNOWLEDGMENTS
Dr. Lanctôt's work was supported by the Alzheimer Society of Canada Research Program and is currently partially supported by the Kunin-Lunenfeld Applied Research Unit of Baycrest Centre for Geriatric Care, Toronto, Ontario.

FIGURE 1. Observations supporting the putative role of 5-HT in the behavioral and psychological symptoms associated with Alzheimer's disease
 |
 |
1 Stoppe G, Brandt CA, Staedt JH: Behavioural problems associated with dementia: the role of newer antipsychotics. Drugs Aging 1999; 14:41–54Crossref, Medline, Google Scholar
2 Mega MS, Cummings JL, Fiorello T, et al: The spectrum of behavioral changes in Alzheimer's disease. Neurology 1996; 46:130–135Crossref, Medline, Google Scholar
3 Hope T, Keene J, Fairburn CJ, et al: Natural history of behavioural changes and psychiatric symptoms in Alzheimer's disease: a longitudinal study. Br J Psychiatry 1999; 174:39–44Crossref, Medline, Google Scholar
4 Reisberg B, Borenstein J, Salob SP, et al: Behavioral symptoms in Alzheimer's disease: phenomenology and treatment. J Clin Psychiatry 1987; 48(suppl 5):9–15Google Scholar
5 Nagaratnam N, Lewis-Jones M, Scott D, et al: Behavioral and psychiatric manifestations in dementia patients in a community: caregiver burden and outcome. Alzheimer Dis Assoc Disord 1998; 12:330–334Crossref, Medline, Google Scholar
6 Cohen CA, Gold DP, Shulman KI, et al: Factors determining the decision to institutionalize dementing individuals: a prospective study. Gerontologist 1993; 33:714–720Crossref, Medline, Google Scholar
7 Merriam AE, Aronson MK, Gaston P, et al: The psychiatric symptoms of Alzheimer's disease. J Am Geriatr Soc 1988; 36:7–12Crossref, Medline, Google Scholar
8 Walsh JS, Welch HG, Larson EB: Survival of outpatients with Alzheimer-type dementia. Ann Intern Med 1990; 113:429–434Crossref, Medline, Google Scholar
9 Rubin EH, Drevets WC, Burke WJ: The nature of psychotic symptoms in senile dementia of the Alzheimer type. J Geriatr Psychiatry Neurol 1988; 1:16–20Crossref, Medline, Google Scholar
10 Lawlor BA, Radcliffe J, Molchan SE, et al: A pilot placebo-controlled study of trazodone and buspirone in Alzheimer's disease. Int J Geriatr Psychiatry 1994; 9:55–59Crossref, Google Scholar
11 Siever LJ, Kahn RS, Lawlor BA, et al: Critical issues in defining the role of serotonin in psychiatric disorders. Pharmacol Rev 1991; 43:509–525Medline, Google Scholar
12 Dubovsky SL, Thomas M: Serotonergic mechanisms and current and future psychiatric practice. J Clin Psychiatry 1995; 56(suppl 2):38–48Google Scholar
13 Fuller RW: Role of serotonin in therapy of depression and related disorders. J Clin Psychiatry 1991; 52(suppl 5):52–57Google Scholar
14 McCormick DA, Williamson A: Convergence and divergence of neurotransmitter action in human cerebral cortex. Proc Natl Acad Sci USA 1989; 86:8098–8102Google Scholar
15 Bowen DM, Allen SJ, Benton JS, et al: Biochemical assessment of serotonergic and cholinergic dysfunction and cerebral atrophy in Alzheimer's disease. J Neurochem 1983; 41:266–272Crossref, Medline, Google Scholar
16 Adolffson R, Gottfries CG, Roos BE, et al: Changes in brain catecholamines in patients with dementia of Alzheimer type. Br J Psychiatry 1979; 135:216–233Crossref, Medline, Google Scholar
17 Arai H, Kosaka K, Iiziku R: Changes of biogenic amines and their metabolites in postmortem brains from patients with Alzheimer-type dementia. J Neurochem 1984; 43:388–393Crossref, Medline, Google Scholar
18 Hardy J, Cowburn R, Barton A, et al: A disorder of cortical GABAergic innervation in Alzheimer's disease. Neurosci Lett 1987; 73:192–196Crossref, Medline, Google Scholar
19 Palmer AM, Stratman GC, Procter AW, et al: Possible neurotransmitter basis of behavioral changes in Alzheimer's disease. Ann Neurol 1988; 23:616–620Crossref, Medline, Google Scholar
20 Nazarali AJ, Reynolds GP: Monoamine neurotransmitters and their metabolites in brain regions in Alzheimer's disease: a postmortem study. Cell Mol Neurobiol 1992; 12:581–587Crossref, Medline, Google Scholar
21 Mann DMA, Yates PO: Serotonin nerve cells in Alzheimer's disease. J Neurol Neurosurg Psychiatry 1983; 46:96–98Crossref, Medline, Google Scholar
22 Yamamoto T, Hirano A: Nucleus raphe dorsalis in Alzheimer's disease: neurofibrillary tangles and loss of large neurons. Ann Neurol 1985; 17:573–7Crossref, Medline, Google Scholar
23 Cross AJ, Crow TJ, Ferrier IN, et al: Serotonin receptor changes in dementia of the Alzheimer type. J Neurochem 1984; 43:1574–1581Google Scholar
24 Francis PT, Palmer AM, Sims NR, et al: Neurochemical studies of early onset Alzheimer's disease. N Engl J Med 1985; 313:7–11Crossref, Medline, Google Scholar
25 Palmer AM, Francis PT, Benton JS, et al: Presynaptic serotonergic dysfunction in patients with Alzheimer's disease. J Neurochem 1987; 48:8–15Crossref, Medline, Google Scholar
26 Chen CPL-H, Alder JT, Bowen DM, et al: Presynaptic serotonergic markers in community-acquired cases of Alzheimer's disease: correlations with depression and neuroleptic medication. J Neurochem 1996; 66:1592–1598Google Scholar
27 Hoogendijk WJG, Feenstra MGP, Botterblom MHA, et al: Increased activity of surviving locus ceruleus neurons in Alzheimer's disease. Ann Neurol 1999; 45:82–91Crossref, Medline, Google Scholar
28 Volicer L, Direnfeld LK, Freedman M, et al: Serotonin and 5-hydroxyindoleacetic acid in CSF: difference in Parkinson's disease and dementia of the Alzheimer's type. Arch Neurol 1985; 42:127–129Crossref, Medline, Google Scholar
29 Murphy DL, Andrews AM, Wichems CH, et al: Brain serotonin neurotransmission: an overview and update with an emphasis on serotonin subsystem heterogeneity, multiple receptors, interactions with other neurotransmitter systems, and consequent implications for understanding the actions of serotonergic drugs. J Clin Psychiatry 1998; 59(suppl 15):4–12Google Scholar
30 Crow TJ, Cross A, Cooper S, et al: Neurotransmitter receptors and monoamine metabolites in the brains of patients with Alzheimer-type dementia and depression, and suicides. Neuropharmacology 1984; 23:1561–1569Google Scholar
31 Middlemiss DN, Palmer AM, Edel N, et al: Binding of the novel serotonin agonist 8-hydroxy-2-(di-n-propylamino) tetralin in normal and Alzheimer brain. J Neurochem 1986; 46:993–996Crossref, Medline, Google Scholar
32 Proctor AW, Middlemiss DN, Bowen DM: Selective loss of serotonin recognition sites in the parietal cortex in Alzheimer's disease. Int J Geriatr Psychiatry 1988; 3:37–44Crossref, Google Scholar
33 Brown GL, Ebert MH, Goyer PE, et al: Aggression, suicide, and serotonin: relationships to CSF amine metabolites. Am J Psychiatry 1982; 139:741–746Crossref, Medline, Google Scholar
34 Perry EK, Perry RH, Candy JM, et al: Cortical serotonin-S2 receptor binding abnormalities in patients with Alzheimer's disease: comparisons with Parkinson's disease. Neurosci Lett 1984; 51:353–357Crossref, Medline, Google Scholar
35 Jansen KL, Faull RL, Dragunow M, et al: Alzheimer's disease: changes in hippocampal N-methyl-d-aspartate, quisqualate, neurotensin, adenosine, benzodiazepine, serotonin and opioid receptors: an autoradiographic study. Neuroscience 1990; 39:613–27Crossref, Medline, Google Scholar
36 Blin J, Baron JC, Dubois B, et al: Loss of brain 5-HT2 receptors in Alzheimer's disease: in vivo assessment with positron emission tomography and [18F] setoperone. Brain 1993; 166(pt 3):497–510Google Scholar
37 Cross AJ, Crow TJ, Ferrier IN, et al: The selectivity of the reduction of serotonin S2 receptors in Alzheimer-type dementia. Neurobiol Aging 1986; 7:3–7Crossref, Medline, Google Scholar
38 Dewar D, Braham DI, McCulloch J:5-HT2 receptors in dementia of Alzheimer type: a quantitative autoradiographic study of frontal cortex and hippocampus. J Neural Transm—Parkinson's Disease and Dementia 1990; 2:129–137Google Scholar
39 Holmes C, Arranz MJ, Powell JF, et al:5-HT2A and 5-HT2C receptor polymorphisms and psychopathology in late onset Alzheimer's disease. Hum Mol Genet 1998; 7:1507–1509Google Scholar
40 Barnes NM, Costall B, Naylor RJ, et al: Normal densities of 5-HT3 receptor recognition sites in Alzheimer's disease. Neuroreport 1990; 1:253–254Crossref, Medline, Google Scholar
41 Weinstein D, Magnuson D, Lee J: Altered G-protein coupling of a frontal cortical low affinity [3H]8-hydroxy-N,N-dipropyl-2-aminotertralin serotonergic binding site in Alzheimer's disease. Behav Brain Res 1996; 73:325–329Crossref, Medline, Google Scholar
42 O'Neill C, Cowburn RF, Wiehager B, et al: Preservation of 5-hydroxytryptamine1A receptor-G protein interactions in the cerebral cortex of patients with Alzheimer's disease. Neurosci Lett 1991; 133:15–19Crossref, Medline, Google Scholar
43 Crews FT, Kurian P, Freund G: Cholinergic and serotonergic stimulation of phosphoinositide hydrolysis is decreased in Alzheimer's disease. Life Sci 1994; 55:1993–2002Google Scholar
44 Marcusson JO, Alafuzoff I, Backstrom IT, et al:5-Hydroxytryptamine-sensitive [3H]imipramine binding of protein nature in the human brain, II: effect of normal aging and dementia disorders. Brain Res 1987; 425:137–145Google Scholar
45 Wallin A, Alafuzoff I, Carlsson A, et al: Neurotransmitter deficits in a non-multi-infarct category of vascular dementia. Acta Neurol Scand 1989; 79:397–406Crossref, Medline, Google Scholar
46 Hansson G, Alafuzoff I, Winblad B, et al: Intact brain serotonin system in vascular dementia. Dementia 1996; 7:196–200Medline, Google Scholar
47 Sparks DL, Markesbery WR: Altered serotonergic and cholinergic synaptic markers in Pick's disease. Arch Neurol 1991; 48:796–799Crossref, Medline, Google Scholar
48 Cheng AV, Ferrier IN, Morris CM, et al: Cortical serotonin-S2 receptor binding in Lewy body dementia, Alzheimer's and Parkinson's diseases. Journal of the Neurological Sciences 1991; 106:50–55Crossref, Medline, Google Scholar
49 Perry EK, Marshall E, Kerwin J, et al: Evidence of a monoaminergic-cholinergic imbalance related to visual hallucination in Lewy body dementia. J Neurochem 1990; 55:1454–1456Google Scholar
50 Lawlor BA, Sunderland T, Mellow AM, et al: A preliminary study of the effects of intravenous m-chlorophenylpiperazine, a serotonin agonist, in elderly subjects. Biol Psychiatry 1989; 25:697–686Crossref, Medline, Google Scholar
51 Lawlor BA, Sunderland T, Mellow AM, et al: Hyperresponsivity to the serotonin agonist m-chlorophenylpiperazine in Alzheimer's disease. Arch Gen Psychiatry 1989; 46:542–549Crossref, Medline, Google Scholar
52 McLoughlin DM, Lucey JV, Dinan TG: Central serotonergic hyperresponsivity in late-onset Alzheimer's disease. Am J Psychiatry 1994; 151:1701–1703Google Scholar
53 Zubenko GS, Moossy J, Martinez AJ, et al: Neuropathologic and neurochemical correlates of psychosis in primary dementia. Arch Neurol 1991; 48:619–624Crossref, Medline, Google Scholar
54 Lawlor BA, Ryan TM, Bierer LM, et al: Lack of association between clinical symptoms and postmortem indices of brain serotonin function in Alzheimer's disease (letter). Biol Psychiatry 1995; 37:895–896Crossref, Medline, Google Scholar
55 Bråne G, Gottfries CG, Blennow K, et al: Monoamine metabolites in cerebrospinal fluid and behavioral ratings in patients with early and late onset of Alzheimer dementia. Alzheimer Dis Assoc Disord 1989; 3:148–156Crossref, Medline, Google Scholar
56 Procter AW, Francis PT, Stratmann GC, et al: Serotonergic pathology is not widespread in Alzheimer patients without prominent aggressive symptoms. Neurochemical Res 1992; 17:917–922Crossref, Medline, Google Scholar
57 Schneider LS, Severson JA, Chui HC, et al: Platelet tritiated imipramine binding and MAO activity in Alzheimer's disease patients with agitation and delusions. Psychiatry Res 1988; 25:311–322Crossref, Medline, Google Scholar
58 Suranyi-Cadotte BE, Gauthier S, Lafaille F, et al: Platelet 3H-imipramine binding distinguishes depression from Alzheimer dementia. Life Sci 1985; 37:2305–2311Google Scholar
59 Mann JJ, McBride PA, Anderson GM, et al: Platelet and whole blood serotonin content in depressed inpatients: correlations with acute and life-time psychopathology. Biol Psychiatry 1992; 32:243–257Crossref, Medline, Google Scholar
60 Mintzer J, Mintzer-Brawman O, Mirski DF, et al: Fenfluramine challenge test as a marker of serotonin activity in patients with Alzheimer's dementia and agitation. Biol Psychiatry 1998; 44:918–921Crossref, Medline, Google Scholar
61 Lanctôt KL, Herrmann N, Eryavec G, et al: Central serotonergic function is related to agitated aggression in Alzheimer's disease (abstract). Clin Pharmacol Ther 1998; 63:222Google Scholar
62 Lanctôt KL, Herrmann N, Eryavec G, et al: Gender variations in effect of sertraline on behavioral disorders (BD) in Alzheimer's disease (AD) (abstract). Clin Pharmacol Ther 1999; 65:190Crossref, Google Scholar
63 Herrmann N, Lanctôt KL: The management of behavioral disturbances in dementia: the role of serotonergic therapies. IDrugs 1998; 1:214–220Medline, Google Scholar
64 Dehlin O, Hedenrud B, Jansson P, et al: A double-blind comparison of alaproclate and placebo in the treatment of patients with senile dementia. Acta Psychiatr Scand 1985; 71:190–196Crossref, Medline, Google Scholar
65 Cutler NR, Haxby J, Kay AD, et al: Evaluation of zimeldine in Alzheimer's disease: cognitive and biochemical measures. Arch Neurol 1985; 42:744–748Crossref, Medline, Google Scholar
66 Auchus AP, Bissey-Black C: Pilot study of haloperidol, fluoxetine, and placebo for agitation in Alzheimer's disease. J Neuropsychiatry Clin Neurosci 1997; 9:591–593Link, Google Scholar
67 Taragano FE, Lyketsos CG, Mangone CA, et al: A double-blind, randomized, fixed-dose trial of fluoxetine vs. amitriptyline in the treatment of major depression complicating Alzheimer's disease. Psychosomatics 1997; 38:246–252Crossref, Medline, Google Scholar
68 Nyth AL, Gottfries CG: The clinical efficacy of citalopram in treatment of emotional disturbances in dementia disorders: a Nordic multicentre study. Br J Psychiatr 1990; 157:894–901Crossref, Medline, Google Scholar
69 Olafsson K, Jorgensen S, Jensen HV, et al: Fluvoxamine in the treatment of demented elderly patients: a double-blind, placebo-controlled study. Acta Psychiatr Scand 1992; 85:453–456Crossref, Medline, Google Scholar
70 Katona CLE, Hunter BN, Bray J: A double-blind comparison of the efficacy and safety of paroxetine and imipramine in the treatment of depression with dementia. Int J Geriatr Psychiatry 1998; 13:100–108Crossref, Medline, Google Scholar
71 Burke WJ, Folks DG, Roccaforte WH, et al: Serotonin reuptake inhibitors for the treatment of coexisting depression and psychosis in dementia of the Alzheimer type. Am J Geriatr Psychiatry 1994; 2:352–354Crossref, Medline, Google Scholar
72 Pollock BG, Mulsant BH, Sweet R, et al: An open pilot study of citalopram for behavioral disturbances of dementia: plasma levels and real-time observations. Am J Geriatr Psychiatry 1997; 5:70–78Crossref, Medline, Google Scholar
73 Swartz JR, Miller BL, Lesser IM, et al: Frontotemporal dementia: treatment response to serotonin selective reuptake inhibitors. J Clin Psychiatry 1997; 58:212–216Crossref, Medline, Google Scholar
74 Hyttel J: Pharmacological characterization of selective serotonin reuptake inhibitors (SSRIs). Int Clin Psychopharmacol 1994; 9(suppl 1):19–26Google Scholar
75 Lee H, Cooney JM, Lawlor BA: The use of risperidone, an atypical neuroleptic, in Lewy body disease. Int J Geriatr Psychiatry 1994; 9:415–417Crossref, Google Scholar
76 Allen RL, Walker Z, D'Ath PJ, et al: Risperidone for psychotic and behavioural symptoms in Lewy body dementia (letter). Lancet 1995; 346(8968):185Google Scholar
77 Jeanblanc W, Davis YB: Risperidone for treating dementia-associated aggression (letter). Am J Psychiatry 1995; 152:1239Medline, Google Scholar
78 Kopala LC, Honer WG: The use of risperidone in severely demented patients with persistent vocalizations. Int J Geriatr Psychiatry 1997; 12:73–77Crossref, Medline, Google Scholar
79 Workman RH Jr, Orengo CA, Bakey AA, et al: The use of risperidone for psychosis and agitation in demented patients with Parkinson's disease. J Neuropsychiatry Clin Neurosci 1997; 9:594–597Link, Google Scholar
80 Geizer M, Ancill RJ: Combination of risperidone and donepezil in Lewy body dementia (letter). Can J Psychiatry 1998; 43:421–422Medline, Google Scholar
81 Orengo CA, Kidwell K, Kunik ME, et al: The effect of risperidone on cognitive performance in elderly psychotic and aggressive patients with dementia; a pilot study. Int J Geriatr Psychopharmacol 1998; 1:193–196Google Scholar
82 Madhusoodanan S, Brenner R, Araujo L, et al: Efficacy of risperidone treatment for psychoses associated with schizophrenia, schizoaffective disorder, bipolar disorder, or senile dementia in 11 geriatric patients: a case series. J Clin Psychiatry 1995; 56:514–518Medline, Google Scholar
83 Herrmann N, Rivard MF, Flynn M, et al: Risperidone for the treatment of behavioral disturbances in dementia: a case series. J Neuropsychiatry Clin Neurosci 1998; 10:220–223Link, Google Scholar
84 Marciniak BH, Guay DRP: Risperidone in the long-term care population. Consultant Pharmacist 1995; 10:1374–1378Google Scholar
85 Frenchman IB, Prince T: Clinical experience with risperidone, haloperidol, and thioridazine for dementia-associated behavioral disturbances. Int Psychogeriatr 1997; 9:431–435Crossref, Medline, Google Scholar
86 Irizarry MC, Ghaemi SN, Lee-Cherry ER, et al: Risperidone treatment of behavioral disturbances in outpatients with dementia. J Neuropsychiatry Clin Neurosci 1999; 11:336–342Link, Google Scholar
87 Jarrett G, Deschenes PJ, Khanna VN: A retrospective study of the effectiveness of risperidone in elderly patients (abstract). Am J Geriatr Psychiatry 1999; 7(4 suppl):44–45Google Scholar
88 Goldberg RJ, Goldberg J: Risperidone for dementia-related disturbed behavior in nursing home residents: a clinical experience. Int Psychogeriatr 1997; 9:65–68Crossref, Medline, Google Scholar
89 Lavretsky H, Sultzer D: A structured trial of risperidone for the treatment of agitation in dementia. Am J Geriatr Psychiatry 1998; 6:127–135Crossref, Medline, Google Scholar
90 Davidson M, Brecher M, and the Risperidone Working Group: Long-term efficacy, safety, and tolerability of risperidone in elderly psychotic patients (abstract). Am J Geriatr Psychiatry 1999; 7(4 suppl):41– 42Google Scholar
91 Katz IR, Jeste DV, Mintzer JE, et al: Comparison of risperidone and placebo for psychosis and behavioral disturbances associated with dementia: a randomized, double-blind trial. Risperidone Study Group. J Clin Psychiatry 1999; 60:107–115Crossref, Medline, Google Scholar
92 De Deyn PP, Rabheru K, Rasmussen A, et al: A randomized trial of risperidone, placebo, and haloperidol for behavioral symptoms of dementia. Neurology 1999; 53:946–955Crossref, Medline, Google Scholar
93 Oberholzer AF, Hendriksen C, Monsch AU, et al: Safety and effectiveness of low-dose clozapine in psychogeriatric patients: a preliminary study. Int Psychogeriatr 1992; 4:187–195Crossref, Medline, Google Scholar
94 Chengappa KNR, Baker RW, Kreinbrook SB, et al: Clozapine use in female geriatric patients with psychoses. J Geriatr Psychiatry Neurol 1995; 8:12–15Medline, Google Scholar
95 Salzman C, Vaccaro B, Lieff J, et al: Clozapine in older patients with psychosis and behavioral disruption. Am J Geriatr Psychiatry 1995; 3:26–33Crossref, Medline, Google Scholar
96 Frankenburg FR, Kalunian D: Clozapine in the elderly. J Geriatr Psychiatry Neurol 1994; 7:129–132Crossref, Medline, Google Scholar
97 Pitner JK, Mintzer JE, Pennypacker LC, et al: Efficacy and adverse effects of clozapine in four elderly psychotic patients. J Clin Psychiatry 1995; 56:180–185Medline, Google Scholar
98 Kinon BJ, Targum SD, Basson BR, et al: Olanzapine in the management of behavioral disturbances and/or psychosis in demented nursing home patients. Presented at 12th Congress, European College of Neuropsychopharmacology, London, September 21–25, 1999Google Scholar
99 Street J, Clark WS, Gannon KS, et al: Olanzapine reduces psychosis and behavioral symptoms associated with Alzheimer's disease (abstract). Am J Geriatr Psychiatry 1999; 7(4 suppl):61– 62Google Scholar
100 Walker Z, Grace J, Overshot R: Olanzapine in dementia with Lewy bodies: a clinical study. Int J Geriatr Psychiatry 1999; 14:459–466Crossref, Medline, Google Scholar
101 McManus DQ, Arvanitis LA, Kowalcyk BB: Quetiapine, a novel antipsychotic: experience in elderly patients with psychotic disorders. Seroquel Trial 48 Study Group. J Clin Psychiatry 1999; 60:292–298Crossref, Medline, Google Scholar
102 Lanctôt KL, Best T, Mittmann N, et al: Efficacy and safety of neuroleptics in behavioural disorders associated with dementia. J Clin Psychiatry 1998; 59:550–561Crossref, Medline, Google Scholar
103 Cantillon M, Brunswick R, Molina D, et al: Buspirone vs. haloperidol: a double-blind trial for agitation in a nursing home population with Alzheimer's disease. Am J Geriatr Psychiatry 1996; 4:263–267Crossref, Medline, Google Scholar
104 Zoldan J, Friedberg G, Goldberg-Stern H, et al: Ondansetron for hallucinations in advanced Parkinson's disease. Lancet 1993; 341:562–563Crossref, Medline, Google Scholar
105 Zoldan J, Friedberg G, Livneh M, et al: Psychosis in advanced Parkinson's disease: treatment with ondansetron, a 5-HT3 receptor antagonist. Neurology 1995; 45:1305–1308Google Scholar
106 Nair NP, Ban TA, Hontela S, et al: Trazodone in the treatment of organic brain syndromes, with special reference to psychogeriatrics. Curr Ther Res Clin Exp 1973; 15:769–775Medline, Google Scholar
107 Simpson DM, Foster D: Improvement in organically disturbed behavior with trazodone treatment. J Clin Psychiatry 1986; 47:191–3Medline, Google Scholar
108 Tingle D: Trazodone in dementia (letter). J Clin Psychiatry 1986; 47:482Medline, Google Scholar
109 Greenwald BS, Marin DB, Silverman SM: Serotoninergic treatment of screaming and banging in dementia (letter). Lancet 1986; 2(8521–8522):1464–1465Google Scholar
110 Pinner E, Rich CL: Effects of trazodone on aggressive behavior in seven patients with organic mental disorders. Am J Psychiatry 1988; 145:1295–6Crossref, Medline, Google Scholar
111 Houlihan DJ, Mulsant BH, Sweet RA, et al: A naturalistic study of trazodone in the treatment of behavioral complications of dementia. Am J Geriatr Psychiatry 1994; 2:78–85Crossref, Medline, Google Scholar
112 Lebert F, Pasquier F, Petit H: Behavioral effects of trazodone in Alzheimer's disease. J Clin Psychiatry 1994; 55:536–538Medline, Google Scholar
113 Sultzer DL, Gray I, Gunay I, et al: A double-blind comparison of trazodone and haloperidol for treatment of agitation in patients with dementia. Am J Geriatr Psychiatry 1997; 5:60–69Crossref, Medline, Google Scholar
114 Petracca G, Teson A, Chemerinski E, et al: A double-blind placebo-controlled study of clomipramine in depressed patients with Alzheimer's disease. J Neuropsychiatry Clin Neurosci 1996; 8:270–275Link, Google Scholar
115 Holzer JC, Gitelman DR, Price BH: Efficacy of buspirone in the treatment of dementia with aggression (letter). Am J Psychiatry 1995; 152:812Medline, Google Scholar
116 Levy MA, Burgio LD, Sweet RA, et al: A trial of buspirone for the control of disruptive behaviors in community-dwelling patients with dementia. Int J Geriatr Psychiatry 1994; 9:841–848Crossref, Google Scholar
117 Herrmann N, Eryavec G: Buspirone in the management of agitation and aggression associated with dementia. Am J Geriatr Psychiatry 1993; 1:249–253Crossref, Medline, Google Scholar
118 Sakauye KM, Camp CJ, Ford PA: Effects of buspirone on agitation associated with dementia. Am J Geriatr Psychiatry 1993; 1:82–84Crossref, Medline, Google Scholar
119 Hargrave R: Serotonergic agents in the management of dementia and post-traumatic stress disorder. Psychosomatics 1993; 34:461–462Crossref, Medline, Google Scholar
120 Hamner M, Huber M: Patient with progressive dementia and choreoathetoid movements treated with buspirone. J Clin Psychopharmacol 1996; 16:261–262Crossref, Medline, Google Scholar
121 Gualtieri C: Buspirone for the behavior problems of patients with organic brain disorders. J Clin Psychopharmcol 1991; 11:280–281Crossref, Medline, Google Scholar
122 Strauss A: Oral dyskinesia associated with buspirone use in an elderly woman. J Clin Psychiatry 1988; 49:322–323Medline, Google Scholar
123 Tiller JW, Dakis JA, Shaw JM: Short-term buspirone treatment in disinhibition with dementia (letter). Lancet 1988; 2(8609):510Google Scholar
124 Colenda CC: Buspirone in treatment of agitated demented patient (letter; published erratum appears in Lancet 1988, 2[8613]:754). Lancet 1988; 1(8595):1169Google Scholar
125 Bergman I, Brane G, Gottfries CG, Jostell KG, Karlsson I, Svennerholm L: Alaproclate: a pharmacokinetic and biochemical study in patients with dementia of Alzheimer type. Psychopharmacology 1983; 80(3):279–83Google Scholar
126 Sobin P, Schneider L, McDermott H: Fluoxetine in the treatment of agitated dementia (letter). Am J Psychiatry 1989; 146(12):1636Google Scholar
127 Ragneskog H, Eriksson S, Karlsson I, Gottfries CG: Long-term treatment of elderly individuals with emotional disturbances: an open study with citalopram. Int Psychogeriatr 1996; 8:659– 68Crossref, Medline, Google Scholar
128 Volicer L, Rheaume Y, Cyr D: Treatment of depression in advanced Alzheimer's disease using sertraline. J Geriatr Psychiatry Neurol 1994; 7:227–229Crossref, Medline, Google Scholar
129 Burke WJ, Dewan V, Wengel SP, et al: The use of selective serotonin reuptake inhibitors for depression and psychosis complicating dementia. Int J Geriatr Psychiatry 1997; 12:519–25Crossref, Medline, Google Scholar
130 Stanislav SW, Fabre T, Crismon ML, et al: Buspirone's efficacy in organic-induced aggression. J Clin Psychopharmacol 1994; 14:126–30Crossref, Medline, Google Scholar
131 Whitehouse PJ, Price DL, Struble RG, et al: Alzheimer's disease and senile dementia: loss of neurons in the basal forebrain. Science 1982; 215:1237–1239Google Scholar
132 Mash DC, Flynn DD, Potter LT: Loss of M2 muscarinic receptors in the cerebral cortex in Alzheimer's disease and experimental cholinergic denervation. Science 1985; 228:1115–1117Google Scholar
133 Bartus RT, Dean RL III, Beer B, et al: The cholinergic hypothesis of geriatric memory dysfunction. Science 1982; 217:408–417Crossref, Medline, Google Scholar
134 Mann DMA, Yates PO, Marcyniuk B: A comparison of changes in the nucleus basalis and locus in Alzheimer's disease. J Neurol Neurosurg Psychiatry 1984; 47:201–203Crossref, Medline, Google Scholar
135 Rossor MN, Rehfeld JF, Emson PC, et al: Normal concentration of cholecystokinin with reduced choline acetyltransferase activity in senile dementia of the Alzheimer's type. Life Sci 1981; 29:405–410Crossref, Medline, Google Scholar
136 Sims NR, Bowen DM, Allen SJ, et al: Presynaptic cholinergic dysfunction in patients with dementia. J Neurochem 1983; 40:503–509Crossref, Medline, Google Scholar
137 Araujo DM, Lapchak PA, Robitaille Y, et al: Differential alterations of various cholinergic markers in cortical and subcortical regions of human brain in Alzheimer's disease. J Neurochem 1988; 50:1914–1923Google Scholar
138 Coyle JT, Price DL, DeLong MR: Alzheimer's disease: a disorder of cortical cholinergic innervation. Science 1985; 219:1184– 1190Google Scholar
139 Drachman DA, Leavitt J: Human memory and the cholinergic system. Arch Neurol 1974; 30:113–121Crossref, Medline, Google Scholar
140 Sitaram N, Weingartner H, Gillin JC: Human serial learning: enhancement with arecholine and choline and impairment with scopolamine. Science 1978; 201:274–276Crossref, Medline, Google Scholar
141 Safer DJ, Allen RP: The central effects of scopolamine in man. Biol Psychiatry 1971; 3:347–355Medline, Google Scholar
142 Squire LR, Knowlton B, Musen G: The structure and organization of memory. Annu Rev Psychology 1993; 44:453–495Crossref, Medline, Google Scholar
143 Weingartner H, Sitaram N, Gillin JC: The role of the cholinergic nervous system in memory consolidation. Bull Psychonomic Soc 1979; 13:9–11Crossref, Google Scholar
144 Davis KL, Hollister LE, Overall J, et al: Physostigmine: effects on cognition and affect in normal subjects. Psychopharmacology 1976; 51:23–27Crossref, Medline, Google Scholar
145 Dunnett SB: The role and repair of forebrain cholinergic systems in short-term memory: studies using the delayed matching-to-position task in rats. Adv Neurol 1993; 59:53–65Medline, Google Scholar
146 Cummings JL, Back C: The cholinergic hypothesis of neuropsychiatric symptoms in Alzheimer's disease. Am J Geriatr Psychiatry 1998; 6(suppl):S64–S78Google Scholar
147 Perry EK, Irving D, Kerwin J, et al: Cholinergic transmitter and neurotrophic activities in Lewy body dementia: similarity to Parkinson's and distinction from Alzheimer disease. Alzheimer Dis Assoc Disord 1993; 7:69–70Crossref, Medline, Google Scholar
148 Perry EK, Marshall E, Thompson P, et al: Monoaminergic activities in Lewy body dementia: relation to hallucinosis and extrapyramidal features. J Neural Transm—Parkinson's Disease 1993; 6:167–177Crossref, Medline, Google Scholar
149 Reinikainen KJ, Soininen H, Riekkinen PJ: Neurotransmitter changes in Alzheimer's disease: implications to diagnostics and therapy. J Neurosci Res 1990; 27:576–586Crossref, Medline, Google Scholar
150 Beal MF, MacGarvey U, Swartz KJ: Galanin immunoreactivity is increased in the nucleus basalis of Meynert in Alzheimer's disease. Ann Neurol 1990; 28:157–161Crossref, Medline, Google Scholar
151 Bowen DM, White P, Spillane JA, et al: Accelerated ageing or selective neuronal loss as an important cause of dementia? Lancet 1979; 1(8106):11–14Google Scholar
152 Bowen DM, Najlerahim A, Procter AW, et al: Circumscribed changes of the cerebral cortex in neuropsychiatric disorders of later life. Proc Natl Acad Sci USA 1989; 86:9504–9508Google Scholar
153 Oishi M, Mochizuki Y, Yoshihashi H, et al: Laboratory examinations correlated with severity of dementia. Ann Clin Lab Sci 1996; 26:340–345Medline, Google Scholar
154 Procter AW, Middlemiss DN, Bowen DM: Selective loss of serotonin recognition sites in the parietal cortex in Alzheimer's disease. Int J Geriatr Psychiatry 1988; 3:37–44Crossref, Google Scholar
155 Jaim-Etcheverry G, Zieher LM: Coexistence of monoamines in peripheral adrenergic neurones, in Co-Transmission, edited by Cuello AC. London, Macmillan, 1982, pp 189–207Google Scholar
156 Chan-Palay V, Asan E: Alterations in catecholamine neurons of the locus coeruleus in senile dementia of the Alzheimer type and in Parkinson's disease with and without dementia and depression. J Comp Neurol 1989; 287:373–392Crossref, Medline, Google Scholar
157 Robbins TW, Everitt BJ: Central norepinephrine neurons and behavior, in Psychopharmacology: The Fourth Generation of Progress, edited by Bloom FE, Kupfer DJ. New York, Raven, 1995, pp 363–372Google Scholar
158 Bondareff W, Mountjoy CQ, Roth M: Loss of neurons of origin of the adrenergic projection to cerebral cortex (nucleus locus ceruleus) in senile dementia. Neurology 1982; 32:164–168Crossref, Medline, Google Scholar
159 Storga D, Vrecko K, Birkmayer JGD, et al: Monoaminergic neurotransmitters, their precursors and metabolites in brains of Alzheimer patients. Neurosci Lett 1996; 203:29–32Crossref, Medline, Google Scholar
160 Palmer AM, Francis PT, Bowen DM, et al: Catecholaminergic neurones assessed ante-mortem in Alzheimer's disease. Brain Res 1987; 414:365–375Crossref, Medline, Google Scholar
161 Perry EK, Tomlinson BE, Blessed G, et al: Neuropathological and biochemical observations on the noradrenergic system in Alzheimer's disease. J Neurol Sci 1981; 51:279–87Crossref, Medline, Google Scholar
162 Gottfries CG, Adolfsson R, Aquilonius SM, et al: Biochemical changes in dementia disorders of Alzheimer type (AD/SDAT). Neurobiol Aging 1983; 4:261–271Crossref, Medline, Google Scholar
163 Palmer AM, Wilcock GK, Esiri MM, et al: Monoaminergic innervation of the frontal and temporal lobes in Alzheimer's disease. Brain Res 1987; 401:231–238Crossref, Medline, Google Scholar
164 Herregodts P, Bruyland M, De Keyser J, et al: Monoaminergic neurotransmitters in Alzheimer's disease: an HPLC study comparing presenile familial and sporadic senile cases. J Neurol Sci 1989; 92:101–116Crossref, Medline, Google Scholar
165 Bierer LM, Haroutunian V, Gabriel S, et al: Neurochemical correlates of dementia severity in Alzheimer's disease: relative importance of the cholinergic deficits. J Neurochem 1995; 64:749– 760Crossref, Medline, Google Scholar
166 Raskind MA, Peskind ER: Neurobiologic bases of noncognitive behavioral problems in Alzheimer disease. Alzheimer Dis Assoc Disord 1994; 8(suppl 3):54–60Google Scholar
167 Raskind MA, Peskind ER, Halter JB, et al: Norepinephrine and MHPG levels in CSF and plasma in Alzheimer's disease. Arch Gen Psychiatry 1984; 41:343–346Crossref, Medline, Google Scholar
168 Lawlor BA, Bierer LM, Ryan TM, et al: Plasma 3-methoxy-4-hydroxyphenylglycol (MHPG) and clinical symptoms in Alzheimer's disease. Biol Psychiatry 1995; 38:185–188Crossref, Medline, Google Scholar
169 Zubenko GS, Moossy J: Major depression in primary dementia: clinical and neuropathologic correlates. Arch Neurol 1988; 45:1182–1186Google Scholar
170 Zweig RM, Ross CA, Hedreen JC, et al: The neuropathology of aminergic nuclei in Alzheimer's disease. Ann Neurol 1988; 24:233–242Crossref, Medline, Google Scholar
171 Förstl H, Burns A, Luther P, et al: Clinical and neuropathological correlates of depression in Alzheimer's disease. Psychol Med 1992; 22:877–884Crossref, Medline, Google Scholar
172 Zubenko GS, Moossy J, Kopp U: Neurochemical correlates of major depression in primary dementia. Arch Neurol 1990; 47:209–214Crossref, Medline, Google Scholar
173 Vitiello B, Veith RC, Molchan SE, et al: Autonomic dysfunction in patients with dementia of the Alzheimer type. Biol Psychiatry 1993; 34:428–433Crossref, Medline, Google Scholar
174 Russo-Neustadt A, Cotman CW: Adrenergic receptors in Alzheimer's disease brain: selective increases in the cerebella of aggressive patients. J Neurosci 1997; 17:5573–5580Google Scholar
175 Meltzer HY: The importance of serotonin-dopamine interactions in the action of clozapine. Br J Psychiatry 1992; 160(suppl 17):22–29Google Scholar
176 Joyce JN, Murray AM, Hurtig HI, et al: Loss of dopamine D2 receptors in Alzheimer's disease with parkinsonism but not Parkinson's or Alzheimer's disease. Neuropsychopharmacology 1998; 19:472–480Crossref, Medline, Google Scholar
177 Pizzolato G, Chierichetti F, Rossato A, et al: Reduced striatal dopamine receptors in Alzheimer's disease: single photon emission tomography study with the D2 tracer [123I]-IBZM. Neurology 1996; 47:1065–1068Google Scholar
178 Itoh M, Meguro K, Fujiwara T, et al: Assessment of dopamine metabolism in brain of patients with dementia by means of 18F-fluorodopa and PET. Ann Nucl Med 1994; 8:245–251Crossref, Medline, Google Scholar
179 Bierer LM, Knott PJ, Schmeidler JM, et al: Post-mortem examination of dopaminergic parameters in Alzheimer's disease: relationship to noncognitive symptoms. Psychiatry Res 1993; 49:211–217Crossref, Medline, Google Scholar
180 Sweet RA, Pollock BG, Mulsant BH, et al: Prolactin response to neuroleptic challenge in late-life psychosis. Psychopharmacol Bull 1995; 31:651–657Medline, Google Scholar
181 Victoroff J, Zarow C, Mack WJ, et al: Physical aggression is associated with preservation of substantia nigra pars compacta in Alzheimer disease. Arch Neurol 1996; 53:428–434Crossref, Medline, Google Scholar
182 Lopez OL, Kaufer D, Reiter CT, et al: Relationship between CSF neurotransmitter metabolites and aggressive behavior in Alzheimer's disease. Eur J Neurol 1996; 3:153–155Crossref, Google Scholar
183 Sweet RA, Pollock BG, Mulsant BH, et al: Association of plasma homovanillic acid with behavioral symptoms in patients diagnosed with dementia: a preliminary report. Biol Psychiatry 1997; 42:1016–1023Google Scholar
184 Grahame-Smith DG: Serotonin in affective disorders. Int Clin Psychopharmacol 1992; 6(suppl 4):5–13Google Scholar
185 Guidotti I, Corda MG, Wise BC, et al: GABAergic synapses: supramolecular organization and biochemical regulation. Neuropharmacology 1983; 22:1471–1479Google Scholar
186 Zorumski CF, Isenberg KE: Insights into the structure and function of GABA-benzodiazepine receptors: ion channels and psychiatry. Am J Psychiatry 1991; 148:162–173Medline, Google Scholar
187 Eichelman B: Neurochemical basis of aggressive behavior. Psychiatric Annals 1987; 17:371–374Crossref, Google Scholar
188 Yew DT, Li WP, Webb SE, et al: Neurotransmitters, peptides, and neural cell adhesion molecules in the cortices of normal elderly humans and Alzheimer patients: a comparison. Exp Gerontol 1999; 34:117–133Crossref, Medline, Google Scholar
189 Mohanakrishnan P, Fowler AH, Vonsattel JP, et al: An in vitro 1H nuclear magnetic resonance study of the temporoparietal cortex of Alzheimer brains. Exp Brain Res 1995; 102:503–510Crossref, Medline, Google Scholar
190 Lowe SL, Francis PT, Procter AW, et al: Gamma-aminobutyric acid concentration in brain tissue at two stages of Alzheimer's disease. Brain 1988; 111(pt 4):785–799Google Scholar
191 Perry TL, Yong VW, Bergeron C, et al: Amino acids, glutathione, and glutathione transferase activity in the brains of patients with Alzheimer's disease. Ann Neurol 1987; 21:331–336Crossref, Medline, Google Scholar
192 Ellison DW, Beal MF, Mazurek MF, et al: A postmortem study of amino acid neurotransmitters in Alzheimer's disease. Ann Neurol 1986; 20:616–621Crossref, Medline, Google Scholar
193 Arai H, Kobayashi K, Ichimiya Y, et al: A preliminary study of free amino acids in the postmortem temporal cortex from Alzheimer-type dementia patients. Neurobiol Aging 1984; 5:319– 321Crossref, Medline, Google Scholar
194 Rossor MN, Garrett NJ, Johnson AL, et al: A post-mortem study of cholinergic and GABA systems in senile dementia. Brain 1982; 105:313–330Crossref, Medline, Google Scholar
195 Sasaki H, Muramoto O, Kanazawa I, et al: Regional distribution of amino acid transmitters in postmortem brains of presenile and senile dementia of Alzheimer type. Ann Neurol 1986; 19:263–269Crossref, Medline, Google Scholar
196 Tarbit I, Perry EK, Perry RH, et al: Hippocampal free amino acids in Alzheimer's disease. J Neurochem 1980; 35:1246–1249Google Scholar
197 Rossor MN, Iversen LL, Reynolds GP, et al: Neurochemical characteristics of early and late onset types of Alzheimer's disease. British Medical Journal (Clin Res Ed) 1984; 288(6422):961– 964Google Scholar
198 Perry EK, Atack JR, Perry RH, et al: Intralaminar neurochemical distributions in human midtemporal cortex: comparison between Alzheimer's disease and the normal. J Neurochem 1984; 42:1402–1410Google Scholar
199 Chu DC, Penney JB Jr, Young AB: Quantitative autoradiography of hippocampal GABAB and GABAA receptor changes in Alzheimer's disease. Neurosci Lett 1987; 82:246–252Crossref, Medline, Google Scholar
200 Chu DC, Penney JB Jr, Young AB: Cortical GABAB and GABAA receptors in Alzheimer's disease: a quantitative autoradiographic study. Neurology 1987; 37:1454–1459Google Scholar
201 Reisine TD, Yamamura HI, Bird ED, et al: Pre- and postsynaptic neurochemical alterations in Alzheimer's disease. Brain Res 1978; 159:477–481Crossref, Medline, Google Scholar
202 Vogt BA, Crino PB, Volicer L: Laminar alterations in gamma-aminobutyric acidA, muscarinic, and beta adrenoceptors and neuron degeneration in cingulate cortex in Alzheimer's disease. J Neurochem 1991; 57:282–290Crossref, Medline, Google Scholar
203 Greenamyre JT, Penney JB, D'Amato CJ, et al: Dementia of the Alzheimer's type: changes in hippocampal l-[3H]glutamate binding. J Neurochem 1987; 48:543–551Crossref, Medline, Google Scholar
204 Carlson MD, Penney JB Jr, Young AB: NMDA, AMPA, and benzodiazepine binding site changes in Alzheimer's disease visual cortex. Neurobiol Aging 1993; 14:343–352Crossref, Medline, Google Scholar
205 Shimohama S, Taniguchi T, Fujiwara M, et al: Changes in benzodiazepine receptors in Alzheimer-type dementia. Ann Neurol 1988; 23:404–406Crossref, Medline, Google Scholar
206 Owen F, Poulter M, Waddington JL, et al: [3H]R05-4864 and [3H]flunitrazepam binding in kainate-lesioned rat striatum and in temporal cortex of brains from patients with senile dementia of the Alzheimer type. Brain Res 1983; 278:373–375Crossref, Medline, Google Scholar
207 Griffiths PD, Perry RH, Crossman AR: A detailed anatomical analysis of neurotransmitter receptors in the putamen and caudate in Parkinson's disease and Alzheimer's disease. Neurosci Lett 1994; 169:68–72Crossref, Medline, Google Scholar
208 Jimenez-Jimenez FJ, Molina JA, Gomez P, et al: Neurotransmitter amino acids in cerebrospinal fluid of patients with Alzheimer's disease. J Neural Transm 1998; 105:269–277Crossref, Medline, Google Scholar
209 Zimmer R, Teelken AW, Trieling WB, et al: Gamma-aminobutyric acid and homovanillic acid concentration in the CSF of patients with senile dementia of Alzheimer's type. Arch Neurol 1984; 41:602–604Crossref, Medline, Google Scholar
210 Manyam NV, Katz L, Hare TA, et al: Levels of gamma-aminobutyric acid in cerebrospinal fluid in various neurologic disorders. Arch Neurol 1980; 37:352–355Crossref, Medline, Google Scholar
211 Weiner MF, Speciale SG, Risser RC, et al: Cerebrospinal fluid and plasma gamma-aminobutyric acid in Alzheimer's disease. Biol Psychiatry 1996; 40:933–934Crossref, Medline, Google Scholar
212 Tohgi H, Abe T, Takahashi S, et al: A selective reduction of excitatory amino acids in cerebrospinal fluid of patients with Alzheimer type dementia compared with vascular dementia of the Binswanger type. Neurosci Lett 1992; 141:5–8Crossref, Medline, Google Scholar
213 Pomara N, Deptula D, Galloway MP, et al: CSF GABA in caregiver spouses of Alzheimer patients. Am J Psychiatry 1989; 146:787–788Crossref, Medline, Google Scholar
214 Bareggi SR, Franceschi M, Bonini L, et al: Decreased CSF concentrations of homovanillic acid and gamma-aminobutyric acid in Alzheimer's disease. Age- or disease-related modifications? Arch Neurol 1982; 39:709–712Google Scholar
215 Enna SJ, Stern LZ, Wastek GJ, et al: Cerebrospinal fluid gamma-aminobutyric acid variations in neurological disorders. Arch Neurol 1977; 34:683–685Crossref, Medline, Google Scholar
216 Achar VS, Welch KM, Chabi E, et al: Cerebrospinal fluid gamma-aminobutyric acid in neurologic disease. Neurology 1976; 26:777–780Crossref, Medline, Google Scholar
217 Sedvall G, Nyback H, Farde L, et al: Neurotransmitter receptor imaging in Alzheimer's disease. J Neural Transm Suppl 1987; 24:43–48Medline, Google Scholar
218 Meyer M, Koeppe RA, Frey KA, et al: Positron emission tomography measures of benzodiazepine binding in Alzheimer's disease. Arch Neurol 1995; 52:314–317Crossref, Medline, Google Scholar
219 Wyper D, Kelly C, Patterson J: Single photon emission computed tomography in Alzheimer's disease: a review of cerebral perfusion and acetylcholine muscarinic receptor imaging and a novel study of the GABA/benzodiazepine system. Int J Geriatr Psychopharmacol 1998; 1:126–133Google Scholar
220 Duman RS: Novel therapeutic approaches beyond the serotonin receptor. Biol Psychiatry 1998; 44:324–335Crossref, Medline, Google Scholar
221 Halliday GM, Li YW, Joh TH, et al: Distribution of substance P-like immunoreactive neurons in the human medulla oblongata: co-localization with monoamine-synthesizing neurons. Synapse 1988; 2:353–370Crossref, Medline, Google Scholar
222 Halliday GM, Li YW, Oliver JR, et al: The distribution of neuropeptide Y-like immunoreactive neurons in the human medulla oblongata. Neuroscience 1988; 26:179–191Crossref, Medline, Google Scholar
223 Gottfries CG, Frederiksen SO, Heilig M: Neuropeptides and Alzheimer's disease. Eur Neuropsychopharmacol 1995; 5:491– 500Crossref, Medline, Google Scholar
224 Minthon L, Edvinsson L, Gustafson L: Correlation between clinical characteristics and cerebrospinal fluid neuropeptide Y levels in dementia of the Alzheimer type and frontotemporal dementia. Alzheimer Dis Assoc Disord 1996; 10:197–203Crossref, Medline, Google Scholar
225 Minthon L, Edvinsson L, Gustafson L: Somatostatin and neuropeptide Y in cerebrospinal fluid: correlations with severity of disease and clinical signs in Alzheimer's disease and frontotemporal dementia. Dement Geriatr Cogn Disord 1997; 8:232– 239Crossref, Medline, Google Scholar
226 de Wied D, van Ree JM: Neuropeptides: animal behaviour and human psychopathology. Eur Arch Psychiatry Neurol Sci 1989; 238:323–331Crossref, Medline, Google Scholar
227 Sargent PA, Bench CJ, Cowen PJ, et al:5-HT1A receptor binding in depressed patients treated with SSRIs using PET and [11C]WAY100635. Neuroimage 1997; 5:A41Google Scholar
228 Drevets WC, Frank E, Price JC, et al: PET imaging of serotonin 1A receptor binding in depression. Biol Psychiatry 1999; 46:1375–1387Google Scholar
229 Sclan SG, Saillon A, Franssen E, et al: The behavior pathology in Alzheimer's disease rating scale (BEHAVE-AD): reliability and analysis of symptom category scores. Int J Geriatr Psychiatry 1996; 11:819–830Crossref, Google Scholar
230 Sparks DL: Aging and Alzheimer's disease: altered cortical serotonergic binding. Arch Neurol 1989; 46:138–40Crossref, Medline, Google Scholar
231 Halliday GM, McCann HL, Pamphlett R, et al: Brain stem serotonin-synthesizing neurons in Alzheimer's disease: a clinicopathological correlation. Acta Neuropathol (Berl) 1992; 84:638–650Crossref, Medline, Google Scholar

